FENOTÍPUS ÉS GENOTÍPUS ELEMZÉSEK RETT SZINDRÓMÁBAN. PhD értekezés tézisei. Dr. Kárteszi Judit. PTE Orvosi Genetikai és Gyermekfejlődéstani Intézet
|
|
|
- Fanni Juhász
- 6 évvel ezelőtt
- Látták:
Átírás
1 ELŐBÍRÁLATI DOLGOZAT FENOTÍPUS ÉS GENOTÍPUS ELEMZÉSEK RETT SZINDRÓMÁBAN PhD értekezés tézisei Dr. Kárteszi Judit PTE Orvosi Genetikai és Gyermekfejlődéstani Intézet Témavezető: Dr. Kosztolányi György egyetemi tanár Programvezető: Dr. Melegh Béla egyetemi tanár
2 TARTALOMJEGYZÉK RÖVIDÍTÉSEK JEGYZÉKE BEVEZETÉS (IRODALMI ÁTTEKINTÉS) 1. A Rett szindróma klinikai jellemzői 2. A Rett szindróma genetikai háttere 3. Fenotípus genotípus elemzés (az X inaktiváció szerepe) 4. Az MeCP2 fehérje vizsgálata VIZSGÁLATI CÉLKITŰZÉSEK 1. A magyarországi Rett szindrómás betegek részletes fenotípus elemzése 2. Az MECP2 mutáció gyakoriságának meghatározása 3. Fenotípus genotípus elemzés 4. Az X inaktiváció szerepének vizsgálata 5. Az MeCP2 fehérje sejten belüli elhelyezkedésének vizsgálata BETEGANYAG ÉS MÓDSZEREK 1. A vizsgált betegpopuláció 2. A fenotípus elemzés módja 3. Az MECP2 mutáció analízise direkt szekvenálással 4. Az X inaktivációs vizsgálat módszere 5. Az immunfluoreszcens vizsgálat leírása EREDMÉNYEK 1. A klinikai vizsgálatok eredményei 2. A mutáció analízis eredményei 3. Fenotípus genotípus elemzés 4. Az X inaktivációs vizsgálat eredményei 5. Az MeCP2 fehérje vizsgálatának eredményei ÖSSZEFOGLALÁS IRODALOM A DOLGOZAT TÉMÁJÁBAN MEGJELENT KÖZLEMÉNYEK ÉS IDÉZHETŐ ABSTRAKTOK MÁS TÉMAKÖRBEN MEGJELENT KÖZLEMÉNYEK ÉS IDÉZHETŐ ABSTRAKTOK KONGRESSZUSI ELŐADÁSOK KÖSZÖNETNYILVÁNÍTÁS 2
3 RÖVIDÍTÉSEK JEGYZÉKE MECP2: methyl CpG kötő fehérje 2 gén MeCP2: methyl CpG kötő fehérje 2 MBD: methyl CpG binding domain TRD: transcriptional repression domain HDAC: histone deacetylase complex EEG: electroencephalogram DEXA: dual energy X ray absorptiometry PCR: polymerase chain reaction CDKL5: cyclin dependent kinase like 5 XIST: X inactivation specific transcript FISH: fluoreszcens in situ hibridizáció UPD: uniparentalis disomia 3
4 BEVEZETÉS (IRODALMI ÁTTEKINTÉS) 1. A Rett szindróma klinikai jellemzői A Rett szindróma idegfejlődési rendellenesség, amelynek gyakorisága irodalmi adatok alapján 1/ / között van. A betegség leírása Andreas Rett nevéhez fűződik, közleménye 1966 ban jelent meg németül (1). Hagberg és munkatársai 1983 ban megjelent angol nyelvű cikke után nagyobb figyelem fordult e különleges betegség felé, amely úgy tűnt, hogy csak leányokat érint (2). A klasszikus Rett szindróma kritériumait 1988 ban nemzetközi megegyezés alapján alkották meg (1. táblázat). 1. táblázat: A Rett szindróma diagnosztikus kritériumai Normális prenatalis és perinatális időszak Látszólag normális pszichomotoros fejlődés az első 6 hónapban Normális születéskori fejkörfogat A fejkörfogatnövekedés lelassulása 5 hónapos és 4 éves kor között A tudatos kézhasználat elmaradása 6 és 30 hónapos kor között Kommunikáció megszünése és átmeneti viselkedésváltozás Súlyos beszédzavar kialakulása Súlyos pszichomotoros elmaradás Sztereotíp kézmozgások (mosó, dörzsölő, tapsoló, ütögető) Végtag és törzsataxia kialakulása 1 és 4 éves kor között A kórkép lefolyása 3 stádiumra osztható: I. Preregresszió: Látszólag normális a fejlődés, de táplálási nehézség, motilitás zavar, figyelemzavar, izomhypotonia előfordul. Nyugodt babák nak írják le őket. Egy két szót elkezdenek mondani, elkezdenek járni, egyedül esznek, majd a fejlődésük megáll, a fejkörfogat növekedése lelassul. 4
5 II. Regresszió: A megszerzett képességeket (beszéd, kézhasználat, interperszonális kapcsolat) gyorsan elveszítik. Mozgáskoordináció és viselkedészavar (autisztikus vonások), sztereotíp kézmozgás, fogcsikorgatás, strabizmus, dystoniás tartás, irregularis légzés alakul ki. A regresszió 8 40 hónapos kor között jelentkezik. III. Postregresszió: A szociabilitás javul, az autisztikus vonások eltűnnek, érzelmileg kötődnek a környezetükhöz. Epilepszia, ízületi deformitás, táplálási nehézség, törzsataxia, lábujjhegyen és széles alapon járás, egyensúlyzavar, tremor, scoliosis, osteoporosis, dystoniás spasmusok, autonom dysfunctio, dyspraxia, alvászavar, agitatív viselkedés, indokolatlan hangulatingadozás jelenik meg. A menarche normális. A fizikális vizsgálat során hypertoniás izomzat, élénk mélyreflexek, atrophiás izomzat, a hossz és súlynövekedés elmaradása, kicsi kezek és lábak észlelhetők. Diszkrét dysmorphiát lehet látni: mélyen ülő szemek, széles orrnyereg, esetleg arcaszimmetria. A hirtelen halál esélye megemelkedett, hátterében feltehetőleg szívarrithmia, illetve agytörzsi diszfunkció áll. A Rett szindróma klinikai megjelenése eltérhet a klasszikus tünet együttestől. Az úgynevezett klasszikus esetek mellett vannak atípusos esetek, lehetnek súlyosabban, vagy éppen ellenkezően, csak kismértékben érintettek a betegek. A Rett szindróma variánsai így magukba foglalják a már korai csecsemőkorban kezdődő, korai epilepsziával járó súlyos formákat és az enyhébb, úgynevezett forme fruste vagy preserved speech variánsokat is (3). 5
6 A Rett szindrómát korábban progresszív betegségnek gondolták, a mai tudás alapján inkább egy alapvetően nem progresszív korai fejlődési zavar, amelynek meghatározott lefolyása van, és amelynek során az élet előrehaladásával különböző szövődmények alakulnak ki. 2. A Rett szindróma genetikai háttere A Rett szindróma X hez kötött domináns módon öröklődik, az esetek többsége sporadikus, de ismertek familiáris esetek is (4). A betegség genetikai okát tekintve számos találgatás látott napvilágot, végül 1999 ben írták le hátterében az X kromoszómán elhelyezkedő methyl CpG kötő fehérje 2 gén (MECP2) mutációit (5). Az MECP2 az X kromoszóma hosszú karján az Xq28 régióban helyezkedik el, és egy ubikviter fehérjét (MeCP2) kódol (1. ábra). 1. ábra: Az X kromoszóma ideogrammja Androgen receptor gén XIST MECP2 6
7 1. A 7
8 A 3 kódoló exonból álló gén két fő funkcionális doménnel rendelkezik. A methyl CpG binding domain (MBD) felelős a metilált DNS hez kötődésért, a transcriptional repression domain (TRD) a histone deacetylase complex (HDAC) felépítésében vesz részt. A leírt mutációk többsége az MBD vagy a TRD valamelyikét érinti. Előfordulnak pontmutációk, inszerciók, kisebb és nagyobb deléciók, illetve komplex átrendeződések is. Egyes mutációk a többinél gyakrabban fordulnak elő. A mutáció vezethet a fehérje egy aminosavának, vagy teljes aminosav sorrendjének megváltozásához, de akár csonka fehérje képződéséhez is. Míg az MBD ban az előbbiek, a TRD ban és a 3 végen az utóbbiak gyakoribbak (6). A különböző tanulmányok a Rett szindrómás betegek kb % ában kimutatták az MECP2 gén valamilyen mutációját (7). A lányoknál jóval ritkábban, fiúknál is leírták már az MECP2 gén mutációit (8). Ezek a betegek általában az atipusos esetek közé sorolhatók. A csekély előfordulási arányt magyarázhatja, hogy az XY nemi kromoszómákat tartalmazó fiú sejtekben a transzkripció kizárólag a mutáns MECP2 génről történik, míg a lányoknál az X inaktiváció a sejtek bizonyos százalékában a károsodott gént érinti, így ezekben a sejtekben a fehérje átíródása az ép allélről történik. Valószínűnek tűnik, hogy a mutáció létrejötte fiúknál gyakran már intrauterin elhalással, esetleg a születést követő egy két éven belüli halállal jár, amikor nem születik diagnózis a nem specifikus tünetek miatt. Egy másik magyarázat, hogy a megfigyelések alapján a mutáció gyakoribb az apai eredetű X kromoszómán, ami a fiú utódoknál hiányzik (9). 8
9 3. Fenotípus genotípus elemzés (az X inaktiváció szerepe) A Rett szindróma klinikai megjelenése, súlyossága széles határok között változik. Adott gyermek fenotípusának meghatározásában, a jelenlegi elképzelések alapján, a mutáció típusán és helyén kívül fontos szerepet játszik az X inaktiváció is. A Lyon hipotézis szerint a női sejtekben jelenlévő két X kromoszóma közül az egyik inaktívvá válik az embrionális élet kezdetén. Az X inaktiváció során minden sejtben az egyik, vagy az apai, vagy az anyai eredetű X inaktiválódik. A véletlen műve, hogy egy adott sejtben melyiket érinti majd a folyamat, de amikor az inaktiválódás bekövetkezett, ennek a sejtnek az összes utódjában ugyanaz az X marad inaktív. A lyonizálódott X en elhelyezkedő gének többsége inaktív, ezért a feltételezések alapján az aktív X en bekövetkező károsodás, mutáció vezethet leginkább a sejt abnormális működéséhez. Ez alapján adódik a felvetés, hogy az X inaktiváció fontos szerepet játszhat több X hez kötött öröklődésű kórkép, így a Rett szindróma fenotípusának kialakításában is. Random X inaktivációról beszélünk, ha az apai vagy az anyai X megközelítőleg fele fele arányban inaktiválódik a sejtekben. Non random (eltolódott) inaktiváció esetén ugyanazon szülőtől származó X inaktiválódik a sejtek több mint 80% ában (a 80 20% os megoszlás elfogadása és általános alkalmazása módszertani okra vezethető vissza). Ha a mutáció heterozigóta formában van jelen, a folyamat vagy a károsodott, vagy az ép allélnek kedvez, és ezzel súlyosbítja, vagy enyhíti a mutáció által meghatározott klinikai képet. 9
10 Az eltolódás lehet a véletlen műve, a sejtek közötti szelekció eredménye, de felvethető az X inaktiváció genetikai meghatározottságának hibája is. Hipotetikus génként az Xq13 on elhelyezkedő XIST et (X inactivation specific transcript) feltételezik, amelynek mutációja az inaktiváció egyenlőtlenné válását eredményezheti. Az X inaktiváció vizsgálata az elmúlt években a figyelem középpontjába került a Rett szindrómával kapcsolatban. Az ebben a témában megjelent cikkek gyakran egymásnak ellentmondó vizsgálati eredményekről számoltak be. Ennek oka nagy valószínűséggel az alacsony esetszám és az eltérő módszerek használata volt. A legújabb irodalmi adatok alapján az X inaktiváció szerepet játszik a Rett szindróma fenotípusának kialakításában. Ismert olyan eset, amikor tünetmentes, vagy csak minimális tüneteket mutató, MECP2 gén mutációt hordozó nőben kimutatták az extrém mértékben eltolódott X inaktivációt (10). Hasonló mechanizmus állhat egyes enyhébb formájú atípusos esetek hátterében is. Több cikkben is közölték, hogy a Rett szindrómásokban, csakúgy, mint más X hez kapcsolódó betegségeknél, gyakrabban fordul elő eltolódott X inaktiváció, mint a normál populációban, leírtak 43% os arányt is (11). Különösen egyes mutáció típusokhoz kapcsolódik gyakrabban non random inaktiváció. 4. Az MeCP2 fehérje vizsgálata A betegség leírása, genetikai diagnosztikája mellett egyre nagyobb erőfeszítések történnek a kezelés irányában. Erre a lehetőséget az adhatja, ha a kóros fehérje szerepe a pathogenezisben tisztázódik. 10
11 Az MeCP2 a metilált DNS hez kötődve kulcsszerepet játszik az úgynevezett transzkripciós silencing komplex kialakításában (12). A komplex feladata a hiszton deacetilálása, amely a kromatin tömörödéséhez vezet. A kromatin ebben a formában elérhetetlenné válik az átíró transzkripciós faktorok számára ( silencing mechanizmusa). Az elméletek szerint az MeCP2 fehérjének jelentős szerepe van a korai agyi fejlődésben. Kevés irodalmi adat áll rendelkezésre a fehérje vizsgálatát illetően. VIZSGÁLATI CÉLKITŰZÉSEK 1. A magyarországi Rett szindrómás betegek részletes fenotípus elemzése Fenotípus genotípus elemzésünk alapja a részletes klinikai megfigyelés, a tünetek leírása és a betegség lefolyásának követése volt. Munkánk során a genetikai vizsgálatokat részletes klinikai vizsgálat előzte meg, amely magába foglalta a részletes anamnézist (kérdőív), fizikális vizsgálatot, röntgen vizsgálatokat, ortopédiai vizsgálatot, pszichológiai és neurológiai vizsgálatot, valamint az EEG elemzést, néhány esetben a cardiológiai és gerinc DEXA vizsgálatot is. Jelen dolgozat az anamnézis és fizikális vizsgálat során nyert információk összefoglalását tartalmazza. 2. Az MECP2 mutáció gyakoriságának meghatározása 11
12 2001 óta végezzük a klinikai vizsgálatok alapján Rett szindrómásnak tartott betegeknél az MECP2 mutáció analízisét. Célul tűztük ki annak megállapítását, hogy a magyarországi Rett szindrómás betegek körében kimutatható e olyan számú esetben mutáció, mint amennyi az irodalmi adatok alapján várható. 3. Fenotípus genotípus elemzés Napjainkban az érdeklődés középpontjába került minden betegségben, így Rett szindrómában is, annak a kérdésnek a megválaszolása, hogy van e direkt összefüggés a klinikai kép és egy adott mutáció (mutáció típus) között, vagy egyéb faktorok is szerepet játszanak a fenotípus kialakításában. 4. Az X inaktiváció szerepének vizsgálata Az irodalmi adatok ismeretében vizsgáltuk, hogy a mutáció analízissel pozitívnak bizonyult magyar Rett szindrómás betegek körében milyen arányban fordul elő eltolódott X inaktiváció, ez milyen kapcsolatban áll a mutáció típusával, és milyen lehetséges hatással van a fenotípus kialakítására. 5. Az MeCP2 fehérje sejten belüli elhelyezkedésének vizsgálata Az MeCP2 fehérje sejten belüli elhelyezkedését vizsgáltuk immunfluoreszcens technikával gyorsan osztódó sejteken (daganatos 12
13 sejtvonal), normális human lymphocytákon és mutációval rendelkező Rett szindrómás betegek lymphocytáin. BETEGANYAG ÉS MÓDSZEREK 1. A vizsgált betegpopuláció 2001 ben kezdtük el a magyarországi Rett szindrómás betegek prospektív klinikai és genetikai vizsgálatát. Először azoknak a betegeknek a szüleit kerestük fel levélben, akiket a Rett Szindróma Társaság számon tartott. Az általuk elküldött címlista szerint ez 47 beteget jelentett, közülük 22 en jelentkeztek levelünkre. A későbbiekben több fórumon beszámoltunk az első eredményekről, amelyet követően Magyarország minden tájáról érkeztek betegek, akiknél a Rett szindróma gyanúja merült fel. Törekedtünk arra, hogy minden gyermeket személyesen is megvizsgálhassunk, sok esetben azonban, az ország távolabbi részeiből, csak a vérmintát kaptuk meg. Eddig összesen 66 leány genetikai vizsgálatát végeztük el, 37 betegnél volt lehetőségünk a részletes fenotípus elemzés elvégzésére is. 2. A fenotípus elemzés módja A betegség kialakulásának, lefolyásának pontos leírásához szerkesztettünk egy kérdőívet, amely hat nagyobb részre tagolódott: családi anamnézis, perinatális anamnézis, pszichomotoros fejlődés, szociális fejlődés, korábbi anamnézis, jelen panaszok. Minden betegnél részletes belszervi és neurológiai vizsgálatot végeztünk. 13
14 A fenotípus elemzést irodalmi ajánlások alapján végeztük el (13). A 2. táblázatban foglaltuk össze a vizsgált tüneteket és az alkalmazott pontozási rendszert: összesen 21, a betegségre jellemzőnek tartott tulajdonságot vizsgáltunk, és a súlyosság alapján 0 2 vagy 0 1 pontot adtunk, a maximális összegzett pontszám 39 lehetett. Minél súlyosabb, illetve jellegzetesebb volt egy adott betegnél a klinikai kép, annál magasabb pontszámot kapott. 14
15 2. táblázat: A fenotípus elemzés pontozási rendszere Vizsgált paraméter Pszichomotoros fejlődés Regresszió kezdete Születéskori fejkörfogat Aktuális fejkörfogat Testsúly Scoliosis Sztereotípia Pont 0 Pont 1 Pont 2 normális 6 hónapos korig normális, de regresszió 6 18 hónapos korban normális, de csökkent növekedés percentil lassú már 6 hónapos korban 3 10 percentil van az idő 25 50% ában valamennyi, segítséggel eszik néhány szó Pozitív EEG, de nem igényel kezelést abnormál hideg kezek, lábak gügyögés, szótagok időnként 8 hónapos korban ataxiás 3 percentil operálták az idő 75 95% ában nincs 18 hónapos korban normális, nincs csökkent növekedés 50 percentil 10 percentil nincs soha Beszéd Epilepszia normál, önállóan eszik normál nincs Légzés Microcirculatio normál normál Nyelvkészség valamilyen beszéd nincs 8 hónapos korban normál Kézhasználat Alvászavar Motoros készség (felülés) Járás Járás kezdete Non verbális kommunikáció Izomtónus Izületi kontraktúrák 18 hónapos korban megőrzött, akaratlagos normál nincs hónapos korban jó szemkontaktus enyhén kóros enyhe 15 6 hónapos korban 3 percentil 3 percentil nincs kezelést igényel nem alkalmaztuk nem alkalmaztuk kiáltozás, nincs hangképzés kifejezett nem ül önállóan soha nem járt, vagy elvesztette a képességét 30 hónapos korban, soha nincs szemkontaktus súlyos zavar súlyos
16 Hangulati labilitás nincs van 16 nem alkalmaztuk
17 3. Az MECP2 mutáció analízise direkt szekvenálással A DNS izolálás perifériás vérből kisózással történt (14). Az MECP2 gén 3 kódoló exonjának (2 es, 3 as és 4 es exon) amplifikálásához az irodalomban korábban publikált hat primer párt használtuk (6). A PCR reakciót 50 µl végtérfogatra számolva a következőképpen állítottuk össze: 1X PCR puffer (50 mm KCL; 10 mm Tris HCl; 0.1 % Triton X 100); 1 mm MgCl2; 200 µm a datp, dctp, dttp, dgtp nukleotidok mindegyikéből; 0.04 µm a forward és reverse primerből; 40 ng/µl DNS. A PCR kivitelezése MJR PTC 150 típusú készüléken történt a következő kondíciók szerint: elődenaturálás 2 perc 96 C on, ezt követően 35 cikluson keresztül 1 perc denaturálás 96 C on, 1 perc primer kötés 55 C on és 1 perc extenzió 72 C on, amelyet 5 perc végső extenzió követett 72 C on. A PCR termékeket direkt forward és reverse szekvenálással analizáltuk ABI PRISM 310 automata szekvenálóval, Big Dye terminátor reagensek felhasználásával. 3. Az X inaktivációs vizsgálat módszere A lyonizáció (inaktiváció) folyamatában a DNS hez metilcsoportok kapcsolódnak. Ez teszi lehetővé az X inaktivációs mintázat meghatározását. A két allél elkülönítésére több módszer ismert. A leggyakrabban és általunk is használt technika a humán androgén receptor gén teszt, amely a génben található CAG trinukleotid ismétlődések számának polimorfizmusán alapul. Ha a vizsgált egyén az ismétlődések számát tekintve heterozigóta, az apai és anyai allél hosszúsága eltérő, a PCR termékek poliakrilamid gélen végzett elektroforézissel egymástól elkülöníthetők. Az androgén receptor génben egy metiláció szenzitív enzim (HpaII) hasítási helyei találhatók. Az 17
18 enzimmel végzett emésztéskor a metilcsoportokkal jelölt inaktív szál védett, ép marad, az aktív szál viszont metilcsoportok hiányában elhasad. Az ennek alapján tervezett primerekkel végzett PCR során kizárólag az inaktív szál amplifikálódik, majd a poliakrilamid gélen megjeleníthető a mintázat. Az előhívás történhet radioaktív módszerrel vagy ezüstözéssel. Módszerünk pontos leírása: A DNS izolálás perifériás vérből kisózással történt (14). PCR reakciót végeztünk a gyermek emésztett és emésztetlen DNS ével, valamint mindkét szülő DNS mintájával az alábbiak alapján. A vizsgált gyermek DNS ét HpaII metiláció szenzitív restrikciós endonukleázzal megemésztettük a gyártó (MBI FERMENTAS) ajánlása szerint. A polimeráz láncreakcióhoz használt primer párok: SBMA A: TCCAGAATCTGTTCCAGAGCGTGC, és SBMA B: GCTGTGAAGGTT GCTGTTCCTCAT. A PCR elegyben 50 µ l végtérfogatra a következő oldatok voltak: 1 X PCR puffer (50 mm KCl; 10mM Tris HCl; 0.1% Triton X 100); 1 mm MgCl2; 200 µ M a datp, dctp, dttp, dgtp nukleodtidok mindegyikéből; 0.04 µ M SBMA A primer; 0.04 µ M SBMA B primer; 40ng/µ l DNS. A PCR kivitelezése MJR PTC 150 típusú készüléken történt. A PCR hőprogram a következő volt: 5 perces 95 ºC os elődenaturáció után 30 cikluson keresztül 1 perc denaturálás 96 C on, 1 perc primer kötődés 62 C on és 1 perc extenzió 72 C on, amelyet a program végén 10 perc végső extenzió követett. A detektálás 2% os agaróz gélelektroforézissel és 8% os poliakrilamid gélelektroforézissel, etídium bromidos festéssel és ezüstözéssel történt (15). A módszer nem informatív, ha a két allélen a trinukleotid ismétlődések száma megegyezik, vagy csak kis mértékben tér el egymástól. Random 18
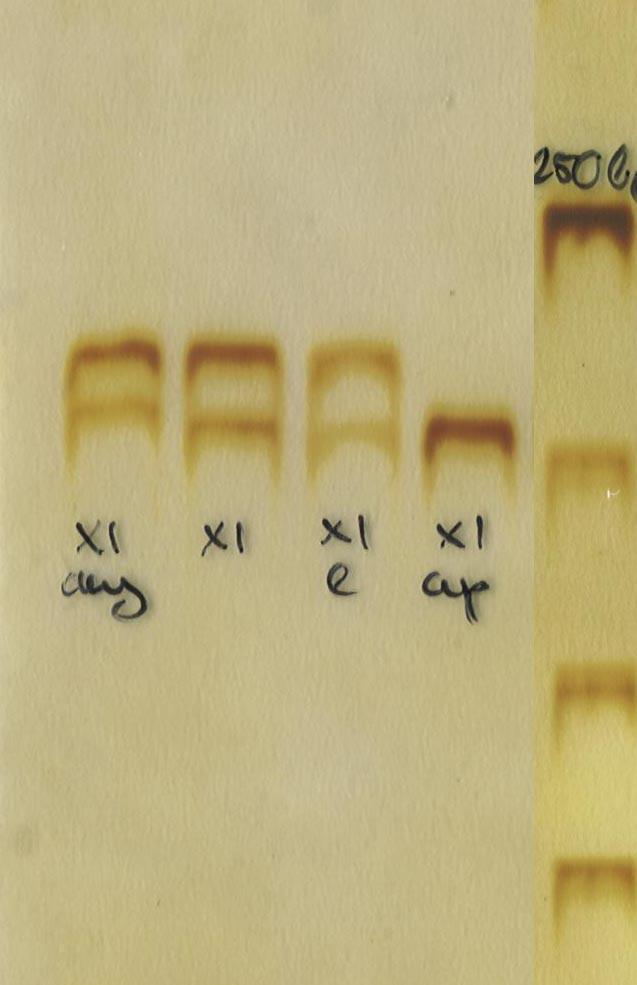
. Non random (eltolódott) inaktivációról beszélünk, ha az emésztett DNS sel végzett PCR mintázatában az egyik allélnek megfelelő band (ez az aktív szál) hiányzik (2. ábra).")

19 inaktivációnak értékeljük a vizsgálatot, ha az emésztetlen DNS en végzett PCR reakció mintázata megegyezik azzal, amit az emésztett DNS minta adott (vagyis mindkét allélnek megfelelő band egyforma erősségű a két reakcióban). Non random (eltolódott) inaktivációról beszélünk, ha az emésztett DNS sel végzett PCR mintázatában az egyik allélnek megfelelő band (ez az aktív szál) hiányzik (2. ábra). Mennyiségi meghatározásra a vizsgálat nem alkalmas, ezért az enyhébb fokú inaktivációk esetleges előfordulását (például 70:30 arányú inaktiváció) ez alapján nem tudjuk megállapítani. 2. ábra: Az X inaktivációs vizsgálat lehetséges eredményei: a, nem informatív; b, non random X inaktiváció; c, random X inaktiváció (ap: apai minta, e: a beteg emésztett mintája, any: anyai minta, nyíl jelöli a beteg emésztetlen mintáját) a, b, 19
20 c, 20
21 4. Az immunfluoreszcens vizsgálat leírása A módszer beállítása REH daganatos sejteken: A REH sejtszuszpenziót (human, leukémiás sejtvonal, pre B sejt) lecentrifugáltuk, a felülúszót leöntöttük, PBS sel mostuk. A fixálás 1% os formaldehiddel szobahőmérsékleten történt (10 perc), majd centrifugáltuk a mintát, a felülúszót leöntöttük. 2 ml PBS t mértünk a sejtekre, majd kettéválasztottuk a mintát, centrifugáltunk, a felülúszót leöntöttük. Az egyikhez hozzáadtuk az anti MeCP2 antitestet (Santa Cruz goat antitest, 1:100 hígítás, 100 μl), a másik volt a kontroll. 37 ºC on inkubáltuk 40 percig, ezt követően PBS sel mostuk. Mindkét csőhöz anti goat FITC jelölt második antitestet adtunk, 37 ºC on inkubáltuk 40 percig. PBS mosást végeztünk, majd az utófixálás 0,1 % os formaldahiddel történt (200 μl). A centrifugálás minden lépésnél 1200 rpm en, 10 percig történt. Human lymphocytákon végzett vizsgálat menete: A sejtek nyerése két módon történt: 1, a heparinos csőbe levett vért Ficollra rétegeztük (1:2 arányban), és 3000 rpm mel, 30 percig centrifugáltuk. A mononuclearis fractiót leszívtuk, fiziológiás sóval mostuk, lecentrifugáltuk a mintát, a felülúszót leöntöttük. 2, egyébként a karyotipizáláshoz használt Chromosome Medium IA táptalajra (GIBCO) 5 csepp vért cseppentettünk, a szuszpenziót 72 órán 21
22 keresztül 37 ºC on inkubáltuk. Lecentrifugáltuk, a felülúszót leöntöttük, és 8 ml 0,05 M KCl os hipotonizáló oldatot mértünk rá, 30 percig 37 ºC on inkubáltuk, majd fiziológiás sóval mostuk. A fixálás, illetve a vizsgálat további menete mindkét esetnél úgy történt, mint a daganatos sejtvonalnál, annyi különbséggel, hogy a mosásokat fiziológiás sóval, nem PBS sel végeztük. EREDMÉNYEK 1. A klinikai vizsgálatok eredményei A fenotípus elemzés eredményeit, a jellemző klinikai tüneteket a 3. táblázatban foglaltuk össze. A Rett szindróma klasszikus definíciója szerint a korai pszichomotoros fejlődés normális, ezért a szerkesztett kérdőív tartalmazta az erre vonatkozó kérdést. Több esetben beszámoltak a szülők arról (a fenotípus elemzésben résztvevő 37 gyermek közül 25 nél), hogy már az első hónapokban jelentkezett valamilyen tünet a következők közül: furcsa testtartás, figyelmetlenség, érdektelenség, tremor, etetési nehézség, súlyfejlődés késés, aluszékonyság, feltűnő nyugodtság, vagy az, hogy a csecsemő nem mosolygott. Egy nemrégen megjelent közlemény szintén hangsúlyozza, hogy vannak korai jelek, amelyek Rett szindrómára utalhatnak (16). A leányok több mint felében a mozgásfejlődés elmaradása már 6 hónapos korban, vagy előtte elkezdődött (57%). A születéskori fejkörfogat minden esetben normális volt, a vizsgálat időpontjában azonban már a betegek 76% ában microcephaliát észleltünk, és nem volt olyan eset, ahol a fejkörfogat elérte volna az 50 percentiles értéket. 14 gyermeknél (38%) 22
23 súlyos dystrophia volt jó étvágy mellett. Nem tartozik a klasszikus tünetek közé, de 15 esetben alacsony növést is észleltünk (40%). Scoliosist 20 betegnél találtunk (54%). 23
24 3. táblázat: A fenotípus elemzés összefoglalása (a klinikai jellemzők gyakoriságát százalékban adtuk meg, kiemeltük azokat, amelyeket a legkarakterisztikusabbnak tartunk) Pszichomotoros fejlődés Kóros (100%). A fejlődés már 6 hónapos korban lassú (57%), vagy ekkor még normális, de regresszió van (43%) Az esetek többségében 6 hónapos kor Regresszió kezdete előtt (57%) vagy 6 18 hónapos kor között (35 %) Születéskori fejkörfogat Normális (100%), de csökkent növekedés (76%) Microcephalia (76%) Aktuális fejkörfogat Dystrophia (38%) Testsúly Fizikális vizsgálattal észlelhető (54%) Scoliosis Sztereotípia Sztereotíp kézmozgás valamilyen formája (100%) A betegek többségében valamilyen Kézhasználat szinten fennmaradt (62%) Beszéd Beszédfejlődés kezdete 1 éves kor körül normális, majd regresszió (100%) Manifeszt convulsiók (49%) Epilepszia Szapora légzés, légzésvisszatartás és Légzés /vagy levegőnyelés (56%) Hideg kezek és/vagy lábak (73%) Microcirculatio Gügyögés, kiabálás (86%) Nyelvkészség A betegek többségében nem volt Alvászavar (83%), viszont a betegség korai fázisában jellemző Többségük tud ülni (68%) Motoros készség (felülés) Járás Ataxiás vagy nem tud járni (100%) Megkésett (83%) Járás kezdete Valamilyen formában, pl. Non verbális kommunikáció szemkontaktus (76%) Valamilyen fokú eltérés (81%) Izomtónus Főleg az Achilles ín kötöttsége (78%) Izületi kontraktúrák Leggyakrabban indokolatlannak tűnő Hangulati labilitás nevetés vagy izgatottság (57%) Érzelmek kifejezése, érzelmek Megőrzött (97%) a súlyos szellemi elmaradás mellett megértése 24

, azonban más fajta kézmozgás is megjelenhet (pl. kezüket szájukba veszik, vagy kezükkel szájukat ütögetik).")

25 A betegség kardinális tünetének számító sztereotíp kézmozgást minden betegünknél megfigyeltük, az esetek többségében a kézmozgás állandó volt. A kézmozgás típusa legtöbbször jellemző, kitekert csuklóval dörzsölik össze kezeiket (3. ábra), azonban más fajta kézmozgás is megjelenhet (pl. kezüket szájukba veszik, vagy kezükkel szájukat ütögetik). Adataink szerint a sztereotíp kézmozgás átlagosan 3 éves kor körül kezdődik (8 hó 6 év), tehát a legtöbb esetben nem tartozik az igazán korai jelek közé. 3. ábra: A Rett szindrómára jellegzetes sztereotíp kézmozgás A betegség másik klasszikus jelének számít, hogy a tudatos kézhasználat elveszik, azonban betegeink 62% ában megfigyelhető volt 25
26 valamilyen szintű kézhasználat (segítséggel eszik, tárgyért nyúl, tárgyat rövid ideig megfog). A betegek 38 % a balkezét részesítette előnyben. Megfigyelésünk szerint leginkább a beszédfejlődésre jellemző a betegség karakterisztikus jegyének tartott normális fejlődés, majd regresszió. Betegeink többsége 1 éves korában elkezdett szavakat mondani, majd átlagosan 1,5 2 éves korban a fejlődés megtorpant és visszafejlődött, néhány betegnél maradt csak meg néhány szó mondása. A legtöbb esetben azonban csak kiabálás, gügyögés volt jellemző (86%). A beszédfejlődés e jellegzetes megtorpanása a Rett szindróma egyik korai jelének tekinthető. Manifeszt epilepszia 18 leánynál volt (49%), általában 4 5 éves életkorban kezdődött, 7 esetben azonban 2 éves kor előtt lépett fel. Légzészavart (szapora légzést, légzésvisszatartást vagy levegőnyelést) az esetek 56% ában észleltünk. Jellemző tünetként említhető a microcirculatio zavara (a kezek és/vagy lábak hidegek voltak a betegek 73% ában), és az izületi kontraktúra, főleg az Achilles ín kötöttsége (78%). Izomtónus eltérést a legtöbb esetben észleltünk (81%), ez leggyakrabban törzs közeli hypotoniát, és a végtagokban fokozott tónust (felső végtagokban fogaskerék tünetet) jelentett. Az esetek egy részében a mélyreflexek élénkek voltak, de kóros reflexet nem észleltünk. Alvászavar a betegek többségében (83%) nem volt, leginkább a regresszió időszakára lehet jellemző, a viselkedésváltozással együtt. Vizsgálataink alapján a sztereotíp kézmozgás és a beszédfejlődés megtorpanása mellett a Rett szindróma legjellemzőbb tünete a járás zavara. A járás késik vagy a gyermek nem tanul meg járni (83%). Ataxiás járást észleltünk a járni tudó gyermekek 100% ában. Felülni a leányok 32% a nem tudott. 26
27 A mentális elmaradással járó betegségek körében különlegesnek számít a Rett szindróma azon vonása, hogy a gyermekek érzelmileg kötődnek környezetükhöz, érzelmeiket kifejezik, környezetük érzéseit megértik (1 beteg kivételével minden gyermek szülei erről számoltak be). Eredeti fenotípus elemzésünk ezt a jellemzőt nem tartalmazta, mert az irodalmi ajánlásokban nem szerepelt. Vizsgálataink alapján azonban ezt a vonást a betegségre jellemzőnek gondoljuk (a fenotípus pontozási rendszert ezzel a jellemzővel a jövőben kiegészítjük). A Rett szindrómás gyermekek kívánságaikat a legtöbb esetben kifejezik a non verbális kommunikáció eszközeivel (pl. szemkontaktus). Gesztusokat viszont nem utánoznak. Hangulati labilitás az esetek 57% ában volt észlelhető. A fenotípus pontozás értékelésekor az életkort fontos figyelembe venni, hiszen vannak olyan tünetek, amelyek idősebb életkorban jelennek csak meg (pl. scoliosis, epilepsia, érzelmi kötődés), és vannak olyanok, amelyek kisgyermekkorban jellemzők (pl. alvászavar, viselkedésváltozás, szemkontaktus kerülése). Az általunk vizsgált gyermekeknél azt láttuk, hogy bár némely esetben az állapot éveken keresztül stagnált vagy kissé javult, a legtöbb gyermeknél fokozatos leromlás volt jellemző a mozgás és kommunikáció terén (11 beteg kontroll vizsgálatát végeztük el 2,5 3 évvel az első vizsgálat után). Betegeinket 6 éves kor alatti és feletti korcsoportra osztottuk (decimális életkort számítottunk). A legfiatalabb beteg 1,67, a legidősebb 21,24 éves volt (3. táblázat). A fenotípus pontozáskor kapott legalacsonyabb pontszám 11, legmagasabb 33 volt. A 6 évesnél fiatalabb betegek átlag pontszáma valamivel alacsonyabb volt (20,4), mint a 6 évnél idősebb betegek átlag pontszáma (23,2). 27
28 A fenotípus elemzésben résztvevő betegeket a Rett szindróma klasszikus formájába sorolhatjuk, bár az egyes esetek lefolyása egyedi különbségeket mutatott. Az egyik gyermeknél például, aki az első vizsgálatkor 12,71 éves volt (22 es eset), és ekkor még 40 szavas szókincse volt, azt gondoltuk, hogy preserved speech variáns, de a 2,5 évvel későbbi kontroll vizsgálatkor állapota sokkal súlyosabb volt, már nem tudott beszélni, csak hangadásra volt képes. Egy másik betegünknél, aki 20,42 éves volt (25 ös eset), a betegség klasszikus tünetei megjelentek kisgyermek korában, viszont vizsgálatunkkor az egyik legjobb állapotú betegnek bizonyult, néhány szót tudott mondani és járásképessége is viszonylag jól megtartott volt ( forme fruste variáns?). Ehhez hasonlóan a 6 os, 11 es és 34 es esetekben is a Rett szindróma enyhébb formáját láttuk, mint a többi betegnél. A 8 as esetünknél, aki az első vizsgálatkor 7,63 éves volt, és ekkor súlyos állapotot észleltünk (nem tudott állni, járni, súlyos epilepsziája volt, szemkontaktus sem tartott), a 3 évvel későbbi kontroll állapotjavulást mutatott, érdeklődőbbé vált (veleszületett súlyos forma?). Hasonlóan súlyos formáját láttuk a betegségnek a 24 es esetben. Betegeink között egy ikerpár volt (14 es és 15 ös eset), akiknél a klinikai kép súlyossága jelentősen különbözött egymástól, bár a fenotípus mindkét esetben klasszikus volt. Fenotípus elemzésünk végén említjük meg a dysmorphológiai vizsgálatot, amely során feltűnően karakterisztikus külső jelet nem észleltünk. A leányok többségének kellemes arcvonásai voltak, tekintetük nem utalt arra a súlyos szellemi elmaradásra, amelyet minden esetben megfigyeltünk. Az irodalomból ismert arc aszimmetriát, valamint a kis kezeket és lábakat mi is több betegnél láttuk. Több esetben észleltünk széles, elálló elülső metszőfogakat, amelyről az irodalomban még nem olvastunk. 28
29 Szinte minden leánynál gótikus szájpadot észleltünk, amelyet összefüggésbe hoznak a magzat csökkent intrauterin mozgásaival. A fenotípus elemzés alapján mondhatjuk, hogy minden olyan gyermeknél érdemes a Rett szindróma diagnózisát felvetni, akinél a fenti pontrendszerrel 20 feletti érték jön ki. Amennyiben ennél alacsonyabb pontszám van, de a beszédfejlődés fenn részletezett megtorpanása és ataxiás járás észlelhető, a Rett szindróma diagnózisa valószínű. Bár a sztereotíp kézmozgás a betegség legismertebb vonása, önmagában nem tekinthetjük abszolút specifikus jelnek. A jellemző sztereotíp kézmozgás a betegség korai stádiumában általában még nem észlelhető, és más ok miatt jelentkező szellemi elmaradásban is megfigyelhető hasonló monoton kézmozgás. 29
30 2. A mutáció analízis eredményei Vizsgálataink során eddig 66 leánynál végeztük el az MECP2 gén mutáció analízisét. A klinikai vizsgálatok alapján 42 esetben tartottuk a Rett szindróma diagnózisát megalapozottnak. Ezek közül 32 esetben (~76%) találtuk meg a mutációt. Ez az arány megfelel más szerzők által korábban észlelt százalékoknak. Tizenegy, már korábban leírt mutációt találtunk 24 betegnél: T158M, R255X 4 betegnél; R294X, R133C 3 betegnél; R106W, R168X, és R270X mindegyikét 2 betegnél; P152R, P225R, 806delG és 1157del41 mindegyikét 1 betegnél. Nyolc Rett szindrómás betegünknél detektáltunk korábban még nem közölt mutációt: S134P, T203M, 276insG; 710delG, 1157del32, 1160del7, 1163del35, 1121del191; 1322del9 (4. és 5. ábra). Azokban az esetekben, ahol nem tudtunk kimutatni mutációt az MECP2 2 es, 3 as és 4 es exonjában, felmerül a gén 1 es exonjának eltérése (17), illetve más gének szerepe a betegség kialakításában. A legújabb ismeretek szerint korai epilepsziával járó formánál a CDKL5 génben található mutáció (18). Az irodalom az Angelman szindrómát említi, mint fő differenciál diagnosztikai problémát, amelynek oka, hogy Rett szindrómában is gyakran észlelhető akaratlan nevetés, hangulatingadozás. Minden betegünknél elvégeztük Angelman szindróma irányában a 15 ös kromoszóma specifikus régiójának FISH analízisét és az MECP2 mutáció negatív betegeknél a 15 ös kromoszóma UPD vizsgálatát is, de eltérést nem találtunk. 30
31 31
32 32
33 3. Fenotípus genotípus elemzés A fenotípus elemzésben résztvevő 37 beteg közül 28 nál tudtunk kimutatni mutációt az MECP2 génben (4. táblázat). A részletes fenotípus elemzést összevetve a mutáció analízis eredményével úgy találtuk, nincs direkt összefüggés a mutáció típusa és a klinikai kép között, más tényezőknek is szerepet kell játszaniuk a fenotípus meghatározásában. A részletes elemzés azért néhány jellegzetességet felfedett. A mutációk szempontjából 4 csoportra osztottuk betegeinket (1. nonsense, 2. missense, 3. deléció a 4 es exonban, 4. normál). Elemzésünket az 5. táblázatban foglaltuk össze. A nonsense mutációs esetekben gyakrabban észleltünk dystrophiát, a súlyos microcephalia is jellemző volt. A missense mutáció esetén gyakoribb volt a microcirculatio zavar és a hangulati labilitás. A 4 es exonban lévő mutáció esetén szemészeti probléma (pl. strabizmus) gyakoribb volt, légzészavar szinte mindegyik gyermeknél megjelent, a beszédet mindegyikük teljesen elhagyta. 9 esetben nem tudtunk kimutatni mutációt az MECP2 gén ismert kódoló régiójában, a klinikai kép viszont egyértelműen Rett szindrómára volt jellemző. Ebben a csoportban a legjellemzőbb közös vonás az volt, hogy minden betegnél a mozgásfejlődés nagymértékben megkésett (csak 5 6 éves korukban tanultak meg járni, illetve többségük nem tanult meg). Az esetek többségében, ahol az MECP2 ben mutációt találtunk és a vizsgálat idejében nem tudtak járni, a járásképesség elvesztése későbbi életkorban történt. Ez alól kivétel volt a fenotípus elemzésnél említett két súlyos állapotú beteg (8 as és 24 es), akik nem tanultak meg járni. 33
34 4. táblázat: A fenotípus gentotípus elemzésben résztvevő betegek X Fenotípus Eset Életkor (év) Mutáció típusa inaktivációs pontszám mintázat 1 8,81 C880T (R294X) NR ,75 N ,91 C316T (R106W) R ,35 C397T (R133C) NI ,68 N ,84 C316T (R106W) NR ,05 C455G (P152R) NR ,63 C502T (R168X) NR ,19 C808T (R270X) R ,24 276insG NR ,83 N , del91;1322del R ,46 N ,47 N ,47 N ,32 C880T (R294X) NI ,15 C763T (R255X) NR ,71 C397T (R133C) NR , del41 R ,64 C808T (R270X) NR ,51 C473T (T158M) NI ,71 C880T (R294X) NR , del7 R ,58 T400C (S134P) ,42 C473T (T158M) NR ,67 C608T (T203M) ,88 C473T (T158M) ,14 N ,16 C502T (R168X) NI , del35 NI ,06 N ,76 710delG NI ,81 C763T (R255X) NR ,62 C397T (R133C) NI ,93 806delG ,64 C763T (R255X) ,07 N 24 34
35 NR: nonrandom, R: random, NI: nem informatív, : nem volt minta a szülőktől. Kékkel kiemeltük a 6 éves illetve annál fiatalabb betegeket. Azoknál a betegeknél, akiknél az MECP2 génben nem találtunk mutációt (N) nem végeztünk X inaktivációs vizsgálatot. 35
36 5. táblázat: A Rett szindróma nem obligát, de karakterisztikus jellemzőinek gyakorisága a mutációk csoportosítása szerint Nonsense Missense 6 hónapos kor előtt megkésett pszichomotoros fejlődés Regresszió kezdete 6 18 hónapos kor között Microcephalia Dystrophia Alacsony növés Scoliosis Nincs tudatos kézhasználat Epilepszia Légzészavar Microcirculatio zavara Nem mond szavakat Alvászavar Kifejezett motoros elmaradás (nem tud ülni) Járás kezdete kifejezetten megkésett vagy nem tanult meg Jó non verbális kommunikáció Izomtónus zavara Izületi kontraktúra Hangulati labilitás 36 Deléció a 4 es exonban Normál
37 Összesen
38 4. Az X inaktivációs vizsgálat eredményei Az X inaktivációs vizsgálatot a fenotípus genotípus analízisben résztvevő, mutációval rendelkező beteg (28 eset) közül 23 nál tudtuk elvégezni, a többi esetben nem állt rendelkezésünkre egyik szülőtől sem DNS minta (3. táblázat). Azoknál a betegeknél, akiknél nem találtunk mutációt, nem végeztük el a vizsgálatot. A 23 eset közül 7 nem volt informatív. Az X inaktivációs mintázatot 5 betegnél randomnak (~22%), 11 betegnél non randomnak (~49%) találtuk. Az irodalmi adatok megoszlanak az eltolódott X inaktiváció arányáról. A tanulmányok többsége csak kevés esetben talált emelkedett arányú eltolódott X inaktivációt, Weaving és munkatársai (11) azonban a közelmúltban hasonló arányú eltolódott X inaktivációt (43%) közöltek, mint amelyet vizsgálataink során mi is találtunk. Adataink tehát noha az általunk vizsgált esetek száma elmaradt az említett tanulmányétól megerősítik az X inaktiváció szerepének jelentőségét a Rett szindróma kialakulásában. Az X inaktiváció jelen tudásunk szerint akkor játszódik le, amikor a blastocysta sejtből áll. Normálisan a folyamat random módon történik, a női magzat sejtjeinek felében az apai, másik felében az anyai X kromoszóma inaktiválódik (minden utódsejtben ugyanaz az X kromoszóma inaktiválódik, mint a progenitor sejtben). Amennyiben a zygota olyan sejteket tartalmaz, amelyekben valamilyen kromoszóma rendellenesség van, eddig nem pontosan ismert folyamatok révén döntően azok a sejtek élnek túl, amelyek genetikai állománya normális (esetlegesen mozaik formában lesz jelen a genetikai eltérés). Amennyiben csak néhány normális progenitor sejt volt a blastocystában, a gyermeknél eltolódott X inaktivációt fogunk észlelni, amennyiben több volt a normális progenitor sejt, az X inaktiváció random lesz (19). 38
39 X hez kötött domináns betegségek esetén logikusan adódik a feltételezés, hogy azok a sejtek élnek döntően túl, amelyekben a mutációt tartalmazó allél inaktiválódik. A 11 non random X inaktivációt mutató esetben egy kivételével azt találtunk, hogy az apai X kromoszóma inaktiválódott (91%). Az irodalmi adatok alapján a sporadikus Rett szindrómás esetek többségében a mutáció az apai X kromoszómán található (9). Azoknál a betegeknél, akiknél a 4 es exonban találtunk deléciót, random X inaktivációt láttunk (a kis esetszám miatt azonban ebből következtetést nem tudtunk levonni). Vizsgálataink alapján azt mondhatjuk, hogy az X inaktiváció a Rett szindróma kialakulásában nagy valószínűséggel szerepet játszik, a fenotípus befolyásolásának részletei azonban nem tisztázottak (ugyanúgy észleltünk eltolódott X inaktivációt az enyhe esetekben, mint klasszikus és súlyos formánál). 5. Az MeCP2 fehérje vizsgálatának eredményei A módszer beállítását daganatos sejtvonalon (REH) végeztük el. A pozitív sejtekben (amelyhez az első antitestet hozzáadtuk), a kontrollhoz viszonyítva egyértelmű jelintenzitást észleltünk a sejtmagban. Flow citometriás vizsgálattal el lehetett választani a negatív és pozitív sejtpopulációt (6. ábra). Human lymphocyta sejteken végezve a vizsgálatot sokkal kisebb jelintenzitást észlelünk, a jelek leginkább a marginális heterochromatinnak megfelelően jelentkeztek. Flow citometriás vizsgálattal nem lehetett a negatív és pozitív sejteket elválasztani. Elvégeztük a vizsgálatot 3 napos 39
40 tenyésztésen átesett lymphocyta sejteken is, ekkor a jelintenzitás emelkedését észleltük, de flow citometriával ekkor sem lehetett különbséget detektálni. Mivel a jelintenzitás a tenyésztett sejteken jobb volt, a későbbiekben öt, mutációval rendelkező Rett szindrómás betegnél tenyésztett sejteken végeztük el az immunfluoreszcens vizsgálatot. A feltevésünk szerint azokban a sejtekben, ahol kóros vagy csonka fehérje képződik, nem mutatható ki jelintenzitás a fehérje C terminálisához kötődő antitestet alkalmazva. Vizsgálataink során nem észleltünk különbséget a mutációval rendelkező betegek lymphocytái és a kontroll lymhocyták között. Ennek az eredménynek az okát nem tudjuk, felvetődik az X inaktiváció szerepe, vagy az, hogy a kóros sejtek nem osztódnak. Vizsgálataink során azt a megfigyelést tettük, hogy gyorsan osztódó sejteken az MeCP2 fehérje nagyobb jelintenzitással észlelhető, mint differenciált sejteken. A jövőben azt szeretnénk megvizsgálni, hogy lehet e szerepe e fehérjének a sejtosztódásban. 40

41 6a. ábra: Az MeCP2 fehérje vizsgálata REH sejten 6b. ábra: A pozitív és negatív sejtpopuláció elválasztása flow citometriával 41
42 ÖSSZEFOGLALÁS A szellemi elmaradással járó betegségek nagy részében még tisztázatlan a genetikai háttér. Azoknak a betegségeknek a pontos megismerése, amelyekben igazolódik egy génnek vagy géneknek a mutációja, segíthetnek megfejteni a fenotípus genotípus összefüggés alapvető szabályait, amelyekről ma még csak sejtéseink vannak. A Rett szindróma egy ilyen betegség, X kromoszómán elhelyezkedő génjét 1999 ben fedezték fel. Az eltelt időben több kutatócsoport beszámolt a klinikai kép sajátságairól, a génben található mutációkról, a fehérje funkciójára vonatkozó adatokról. Vizsgálatainkat 2001 ben kezdtük el Magyarországon elsőként, az akkor rendelkezésre álló irodalmi adatok áttekintése után. A következő céljaink voltak: 1. a lehető legrészletesebb klinikai vizsgálat elvégzése (olyan tünetek keresése, amelyek nagy biztonsággal felismerhetővé és más betegségektől megkülönböztethetővé teszik a Rett szindrómát), 2. az MECP2 szekvencia analízisének beállítását követően a klinikailag diagnosztizált esetek mutáció analízisének elvégzése, a mutáció hazai gyakoriságának megállapítása, 3. fenotípus genotípus összefüggés keresése, 4. adatok nyerése a betegség patomechanismusának megértésére. 1. Az elvégzett klinikai vizsgálatok alapján konkrét ajánlást tudunk tenni arra vonatkozóan, hogy milyen klinikai tünetek esetén érdemes elvégezni az MECP2 mutációját. Kidolgoztunk irodalmi ajánlások alapján egy fenotípus pontozási rendszert, amelynek alkalmazásával jó találati arány (50 80% között) biztosítható az MECP2 mutáció analízis során. A betegség egyik korai jele a járás késése illetve bizonytalansága, látszólag normális kezdeti szellemi fejlődés mellett. Karakterisztikus, hogy a 42
43 beszédfejlődés normálisan indul, majd 1,5 éves kor körül megáll és visszafejlődik. A betegségre jellegzetes sztereotíp kézmozgás késői jelnek tekinthető, csak átlagosan 3 éves kor körül jelentkezik. Más szellemi elmaradással járó betegségektől a Rett szindróma különbözik abban a tekintetben, hogy az érzelmi értés és kifejezés fennmarad. 2. A klinikai vizsgálatok alapján 42 esetben tartottuk a Rett szindróma diagnózisát megalapozottnak, ezek közül 32 esetben (~76%) találtuk meg a mutációt. Ez az eredmény megfelel az irodalomban közölt adatoknak (50 80%). Nyolc Rett szindrómás betegünknél korábban még nem közölt mutációt detektáltunk. 3. Az X inaktivációs vizsgálatok során azt találtuk, hogy a Rett szindrómás betegeknél nagy arányban (49%) fordul elő eltolódott (non random) X inaktiváció. Weaving és munkatársai (11) a közelmúltban hasonló arányú eltolódott X inaktivációt (43%) közöltek. Azt is megfigyeltük, hogy 1 beteg esetének kivételével az apai X kromoszóma inaktiválódott (91%). Az irodalmi adatok alapján sporadikus Rett szindrómában a mutáció az esetek többségében az apai X kromoszómán található. A fenotípus genotípus elemzés eredményét összefoglalva úgy tűnik, önmagában sem a mutáció helye, típusa, sem az X inaktiváció nincs direkt összefüggésben a fenotípussal. Egyes esetekben azonban az X inaktiváció valószínűleg befolyásolja a klinikai képet. 4. Az MeCP2 fehérje kimutatására végzett vizsgálatok a dolgozat megírásáig csak kezdeti stádiumig jutottak. Az immunfluoreszcens vizsgálatok során azt láttuk, hogy osztódó sejtekben a jelintenzitás sokkal nagyobb, mint differenciált sejtekben. heterokromatinnak megfelelően láttuk. 43 A jeleket a marginális
44 A betegség mechanizmusának megértéséhez szükségesnek tartjuk e vizsgálatok folytatását. Szeretnénk megérteni a fehérje sejtekben betöltött funkcióját, ennek vizsgálatára Western blott és további immunfluoreszcens vizsgálatokat tervezünk. 44
45 IRODALOM 1. Rett A: On an unusual brain atrophy syndrome in hyperammonemia in childhood. Wien Med Wochenschr, 1966, 116, Hagberg B, Aicardi J, Dias K, Ramos O: A progressive syndrome of autism, dementia, ataxia and loss of purposeful hand use in girls: Rett s syndrome: Report of 35 cases. Ann Neurol, 1983, 14, Zappella M, meloni I, Longo I, Hayek G, Renieri A: Preserved Speech Variants of the Rett Syndrome: Molecular and Clinical Analysis. Am J Med Genet, 2001, 104, Webb T, Latif F: Rett syndrome and the MECP2 gene. J Med Genet, 2001, 38: Amir RE, Van den Veyver IB, Wan M, Tran CQ, Franke U, Zoghbi HY: Rett syndrome is caused by mutations in X linked MECP2, encoding methyl CpG binding protein 2. Nat Genet, 1999, 23: Buyse IM, Fang P, Hoon KT, Amir RE, Zoghbi HY, Roa BB: Diagnostic testing for Rett syndrome by DHPLC and direct sequencing analysis of the MECP2 gene: identification of several novel mutations and polymorphisms. Am J Hum Genet, 2000, 67, Nielsen JB, Henriksen KF, Hansen C, Silahtaroglu a, Schwartz M, Tommerup N: MECP2 mutations in Danish patients with Rett syndrome: high frequency of mutations but no consistent correlations with clinical severity or with the X chromosome inactivation pattern. Eur J Hum Genet, 2001, 9, Villard L, cardoso AK, Chelly PJ, tardieu PM, Fontes M: Two affected boys in a Rett syndrome family: clinical and molacular findings. Neurology, 2000, 55, Girard M, Couvert P, Carrie A, tardieu M, Chelly J, Beldjord C, Bienvenu T: Parnetal origin of de novo MECP2 mutations in Rett syndrome. Eur J Hum Genet, 2001, 9, Villard L, Levy N, Xiang F, Kpebe A, Labelle V, Chevillard C, Zhang Z, Schwartz CE, Tardieu M, Chelly J, Anvret M, Fontes M: Segregation of a totally skewed pattern of X chromosome inactivation in four familial cases of Rett syndrome without MECP2 mutation: implication for the disease. J Med Genet, 2001, 38,
46 11.Weaving LS, Williamson SL, Bennetts B, Davis M, Ellaway CJ, Leonard H, Thong MK, Delatycki M, Thompson EM, Laing N, Christodoulou J: Effects of MECP2 mutation type, location and X inactivation in modulating Rett syndrome phenotype. Am J Med Genet, 2003, 118A, Nan X, Huck Hui Ng, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A: Transcriptional repression by the methyl CpG binding protein MeCP2 involves a histone deacetylase complex. Nature, 1998, 393: Kerr AM, nomura Y, Armstrong D, Anvret M, Belichenko PV, Budden S, cass H, Christodoulou J, Clarke A, Ellaway C, d Esposito M, Francke U, Hulten M, Julu P, Leonard H, Naidu S, Schanen C, Webb T, Engerstrom IW, Yamashita Y, Segawa M: Guidelines for reporting clinical features in cases with MECP2 mutations. Brain and Dev, 2001, 23, Miller SA, dykes DD, Polesky HF: A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acidds Res, 1988, 16, Melegh B, Molnar J: Nonisotopic method for precise detection of (CAG)n repeats. Clin Chem, 1996, 42, Einspieler C, Kerr AM, Prechtl HFR: Is the Early Development of Girls with Rett Disorder Really Normal? Pediatric Research, 2005, 57, Evans JC, Hayley LA, Whatley SD, Kerr A, Clarke A, Butler R: Variation in exon 1 coding region and promoter of MECP2 in Rett syndrome and controls. Eur J Hum Genet, published online 2004 september. 18. Renieri A, Nari F, Azimonti S, Caselli R, Scala E, Longo I, Pesucci C, Ariani F, Broccoli V, Bolognesi F, Bertani I, Zappella M, Kilstru Nielsen C, Landsberger N: CDKL5 interacts with MeCP2 and it is responsible for the early seizure variant of Rett syndrome. Eur J Hum Genet 2005, 13, C Robinson WP: Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays 2000, 22:
47 A DOLGOZAT TÉMÁJÁBAN MEGJELENT KÖZLEMÉNYEK ÉS IDÉZHETŐ ABSTRAKTOK 1. Kárteszi J., Hollódy K., Bene J., Morava É., Hadzsiev K., Czakó M., Melegh B., Kosztolányi Gy.: Az MECP2 gén mutációinak analízise direkt szekvenálással magyarországi Rett szindrómás betegekben. Orvosi Hetilap 2004, 145 (17): Kárteszi J., Hollódy K., Bene J., Morava É., Hadzsiev K., Czakó M., Melegh B., Tészás A., Kosztolányi Gy.: Mutation analysis of MECP2 and determination of the X inactivation pattern in Hungarian Rett syndrome patients. Am J Med Genet 2004, 131 (1): Kárteszi J., Bene J., Morava É, Czakó M., Hollódy K., Melegh B., Kosztolányi Gy.: Analysis of the MECP2 gene by direct sequencing in Hungarian Rett syndrome patients. Eur J Hum Genet 2002, 10, P Weisenbach J., Hollódy K., Kárteszi J., Hadzsiev K, Melegh B., Kosztolányi Gy.: Characteristic X ray sign of Rett syndrome: extreme thin diaphysis with narrow medulla of tubular bones. Eur J Hum Genet 2003, 11, P Kárteszi J., Bene J., Hollódy K., Morava É., Hadzsiev K., Czakó M., Melegh B., Tészás A., Kosztolányi Gy.: Mutation analysis of MECP2 and determination of the X inactivation pattern in Hungarian Rett Syndrome patients. Eur J Hum Genet 2004, 12, P050. MÁS TÉMAKÖRBEN MEGJELENT KÖZLEMÉNYEK IDÉZHETŐ ABSTRAKTOK ÉS 1. Kárteszi J., Morava É., Czakó M., Gáti I., Czopf J., Kosztolányi Gy., Melegh B.: Kennedy betegség egy progresszív beszédzavarban szenvedő férfiben. Orvosi Hetilap 2001, 142 (35): Kárteszi J., Morava É., Czakó M., Kosztolányi Gy.: Smith Magenis szindróma genetikailag kódolt viselkedészavar? Gyermekgyógyászat 2001, 52 (4): Cser B., Morava É., Czakó M., Kárteszi J., Szőnyi L., Wevers R., Kosztolányi Gy.: Veleszületett glikozilációs zavar (CDG Ia betegség) egy izomhipotóniás, hepatopathiás gyermekben. Gyermekgyógyászat 2003, 54 (1):
FENOTÍPUS ÉS GENOTÍPUS ELEMZÉSEK RETT SZINDRÓMÁBAN. PhD értekezés. Dr. Kárteszi Judit. PTE Orvosi Genetikai és Gyermekfejlődéstani Intézet
 FENOTÍPUS ÉS GENOTÍPUS ELEMZÉSEK RETT SZINDRÓMÁBAN PhD értekezés Dr. Kárteszi Judit PTE Orvosi Genetikai és Gyermekfejlődéstani Intézet Témavezető: Dr. Kosztolányi György egyetemi tanár Programvezető:
FENOTÍPUS ÉS GENOTÍPUS ELEMZÉSEK RETT SZINDRÓMÁBAN PhD értekezés Dr. Kárteszi Judit PTE Orvosi Genetikai és Gyermekfejlődéstani Intézet Témavezető: Dr. Kosztolányi György egyetemi tanár Programvezető:
FENOTÍPUS ÉS GENOTÍPUS ELEMZÉSEK RETT SZINDRÓMÁBAN. PhD értekezés. Dr. Kárteszi Judit. PTE Orvosi Genetikai és Gyermekfejlődéstani Intézet
 FENOTÍPUS ÉS GENOTÍPUS ELEMZÉSEK RETT SZINDRÓMÁBAN PhD értekezés Dr. Kárteszi Judit PTE Orvosi Genetikai és Gyermekfejlődéstani Intézet Témavezető: Dr. Kosztolányi György egyetemi tanár Programvezető:
FENOTÍPUS ÉS GENOTÍPUS ELEMZÉSEK RETT SZINDRÓMÁBAN PhD értekezés Dr. Kárteszi Judit PTE Orvosi Genetikai és Gyermekfejlődéstani Intézet Témavezető: Dr. Kosztolányi György egyetemi tanár Programvezető:
A genetikai lelet értelmezése monogénes betegségekben
 A genetikai lelet értelmezése monogénes betegségekben Tory Kálmán Semmelweis Egyetem, I. sz. Gyermekklinika A ~20 ezer fehérje-kódoló gén a 23 pár kromoszómán A kromoszómán található bázisok száma: 250M
A genetikai lelet értelmezése monogénes betegségekben Tory Kálmán Semmelweis Egyetem, I. sz. Gyermekklinika A ~20 ezer fehérje-kódoló gén a 23 pár kromoszómán A kromoszómán található bázisok száma: 250M
Az X kromoszóma inaktívációja. A kromatin szerkezet befolyásolja a génexpressziót
 Az X kromoszóma inaktívációja A kromatin szerkezet befolyásolja a génexpressziót Férfiak: XY Nők: XX X kromoszóma: nagy méretű több mint 1000 gén Y kromoszóma: kis méretű, kevesebb, mint 100 gén Kompenzációs
Az X kromoszóma inaktívációja A kromatin szerkezet befolyásolja a génexpressziót Férfiak: XY Nők: XX X kromoszóma: nagy méretű több mint 1000 gén Y kromoszóma: kis méretű, kevesebb, mint 100 gén Kompenzációs
Molekuláris genetikai vizsgáló. módszerek az immundefektusok. diagnosztikájában
 Molekuláris genetikai vizsgáló módszerek az immundefektusok diagnosztikájában Primer immundefektusok A primer immundeficiencia ritka, veleszületett, monogénes öröklődésű immunhiányos állapot. Családi halmozódást
Molekuláris genetikai vizsgáló módszerek az immundefektusok diagnosztikájában Primer immundefektusok A primer immundeficiencia ritka, veleszületett, monogénes öröklődésű immunhiányos állapot. Családi halmozódást
Prenatalis diagnosztika lehetőségei mikor, hogyan, miért? Dr. Almássy Zsuzsanna Heim Pál Kórház, Budapest Toxikológia és Anyagcsere Osztály
 Prenatalis diagnosztika lehetőségei mikor, hogyan, miért? Dr. Almássy Zsuzsanna Heim Pál Kórház, Budapest Toxikológia és Anyagcsere Osztály Definíció A prenatális diagnosztika a klinikai genetika azon
Prenatalis diagnosztika lehetőségei mikor, hogyan, miért? Dr. Almássy Zsuzsanna Heim Pál Kórház, Budapest Toxikológia és Anyagcsere Osztály Definíció A prenatális diagnosztika a klinikai genetika azon
DNS-szekvencia meghatározás
 DNS-szekvencia meghatározás Gilbert 1980 (1958) Sanger 3-1 A DNS-polimerázok jellemzői 5'-3' polimeráz aktivitás 5'-3' exonukleáz 3'-5' exonukleáz aktivitás Az új szál szintéziséhez kell: templát DNS primer
DNS-szekvencia meghatározás Gilbert 1980 (1958) Sanger 3-1 A DNS-polimerázok jellemzői 5'-3' polimeráz aktivitás 5'-3' exonukleáz 3'-5' exonukleáz aktivitás Az új szál szintéziséhez kell: templát DNS primer
Hátterükben egyetlen gén áll, melynek általában számottevő a viselkedésre gyakorolt hatása, öröklési mintázata jellegzetes.
 Múlt órán: Lehetséges tesztfeladatok: Kitől származik a variáció-szelekció paradigma, mely szerint az egyéni, javarészt öröklött különbségek között a társadalmi harc válogat? Fromm-Reichmann Mill Gallton
Múlt órán: Lehetséges tesztfeladatok: Kitől származik a variáció-szelekció paradigma, mely szerint az egyéni, javarészt öröklött különbségek között a társadalmi harc válogat? Fromm-Reichmann Mill Gallton
MUTÁCIÓK. A mutáció az örökítő anyag spontán, maradandó megváltozása, amelynek során új genetikai tulajdonság keletkezik.
 MUTÁCIÓK A mutáció az örökítő anyag spontán, maradandó megváltozása, amelynek során új genetikai tulajdonság keletkezik. Pontmutáció: A kromoszóma egy génjében pár nukleotidnál következik be változás.
MUTÁCIÓK A mutáció az örökítő anyag spontán, maradandó megváltozása, amelynek során új genetikai tulajdonság keletkezik. Pontmutáció: A kromoszóma egy génjében pár nukleotidnál következik be változás.
Önkéntes némaság - a mutizmus. Írta: Csányi Nikolett
 Mi is a mutizmus? Az alkalmazkodó viselkedés zavara. A mutizmus némaságot jelent. Mutizmus során a beszédszervek épek, de súlyos viselkedéses gátlás (neurotikus zavar) alakul ki, és a gyermek nem beszél.
Mi is a mutizmus? Az alkalmazkodó viselkedés zavara. A mutizmus némaságot jelent. Mutizmus során a beszédszervek épek, de súlyos viselkedéses gátlás (neurotikus zavar) alakul ki, és a gyermek nem beszél.
A stresszteli életesemények és a gyermekkori depresszió kapcsolatának vizsgálata populációs és klinikai mintán
 A stresszteli életesemények és a gyermekkori depresszió kapcsolatának vizsgálata populációs és klinikai mintán Doktori értekezés tézisei Dr. Mayer László Semmelweis Egyetem Mentális Egészségtudományok
A stresszteli életesemények és a gyermekkori depresszió kapcsolatának vizsgálata populációs és klinikai mintán Doktori értekezés tézisei Dr. Mayer László Semmelweis Egyetem Mentális Egészségtudományok
Post-varicella angiopathia (PVA): klinikai és radiológiai jellemzők összefoglalása hét eset alapján
: klinikai és radiológiai jellemzők összefoglalása hét eset alapján") Post-varicella angiopathia (PVA): klinikai és radiológiai jellemzők összefoglalása hét eset alapján Kovács Éva 1,Várallyay György 2, Harkányi Zoltán 1, Rosdy Beáta 3, Móser Judit 3, Kollár Katalin 3, Barsi
Post-varicella angiopathia (PVA): klinikai és radiológiai jellemzők összefoglalása hét eset alapján Kovács Éva 1,Várallyay György 2, Harkányi Zoltán 1, Rosdy Beáta 3, Móser Judit 3, Kollár Katalin 3, Barsi
De novo SNC1A géndeléció terápia rezisztens Dravet szindrómában
 De novo SNC1A géndeléció terápia rezisztens Dravet szindrómában Bene Judit 1,2, Hadzsiev Kinga 1,2, Komlósi Katalin 1, Kövesdi Erzsébet 1,2, Mátyás Petra 1, Melegh Béla 1 1 Pécsi Tudományegyetem, Általános
De novo SNC1A géndeléció terápia rezisztens Dravet szindrómában Bene Judit 1,2, Hadzsiev Kinga 1,2, Komlósi Katalin 1, Kövesdi Erzsébet 1,2, Mátyás Petra 1, Melegh Béla 1 1 Pécsi Tudományegyetem, Általános
In Situ Hibridizáció a pathologiai diagnosztikában és ami mögötte van.
 In Situ Hibridizáció a pathologiai diagnosztikában és ami mögötte van. Kneif Józsefné PTE KK Pathologiai Intézet Budapest 2017. 05. 26 Kromoszóma rendellenesség kimutatás PCR technika: izolált nukleinsavak
In Situ Hibridizáció a pathologiai diagnosztikában és ami mögötte van. Kneif Józsefné PTE KK Pathologiai Intézet Budapest 2017. 05. 26 Kromoszóma rendellenesség kimutatás PCR technika: izolált nukleinsavak
Engedélyszám: 18211-2/2011-EAHUF Verziószám: 1. 2460-06 Humángenetikai vizsgálatok követelménymodul szóbeli vizsgafeladatai
 1. feladat Ismertesse a gyakorlaton lévő szakasszisztens hallgatóknak a PCR termékek elválasztása céljából végzett analitikai agaróz gélelektroforézis során használt puffert! Az ismertetés során az alábbi
1. feladat Ismertesse a gyakorlaton lévő szakasszisztens hallgatóknak a PCR termékek elválasztása céljából végzett analitikai agaróz gélelektroforézis során használt puffert! Az ismertetés során az alábbi
Kutatási beszámoló ( )
") Kutatási beszámoló (2008-2012) A thrombocyták aktivációja alapvető jelentőségű a thrombotikus betegségek kialakulása szempontjából. A pályázat során ezen aktivációs folyamatok mechanizmusait vizsgáltuk.
Kutatási beszámoló (2008-2012) A thrombocyták aktivációja alapvető jelentőségű a thrombotikus betegségek kialakulása szempontjából. A pályázat során ezen aktivációs folyamatok mechanizmusait vizsgáltuk.
11. Dr. House. Biokémiai és sejtbiológiai módszerek alkalmazása az orvoslásban
 11. Dr. House. Biokémiai és sejtbiológiai módszerek alkalmazása az orvoslásban HIV fertőzés kimutatása - (fiktív) esettanulmány 35 éves nő, HIV fertőzöttség gyanúja. Két partner az elmúlt időszakban. Fertőzött-e
11. Dr. House. Biokémiai és sejtbiológiai módszerek alkalmazása az orvoslásban HIV fertőzés kimutatása - (fiktív) esettanulmány 35 éves nő, HIV fertőzöttség gyanúja. Két partner az elmúlt időszakban. Fertőzött-e
Az érzelmi felismerés viselkedészavaros lányokban (Emotion recognition in girls with conduct problems)
") Az érzelmi felismerés viselkedészavaros lányokban (Emotion recognition in girls with conduct problems) Christina Schwenck, Angelika Gensthaler Marcel Romanos Christine M. Freitag, Wolfgang Schneider, Regina
Az érzelmi felismerés viselkedészavaros lányokban (Emotion recognition in girls with conduct problems) Christina Schwenck, Angelika Gensthaler Marcel Romanos Christine M. Freitag, Wolfgang Schneider, Regina
Pszichotrauma és disszociatív kapacitás összefüggésének vizsgálata syncopés betegek körében
 Pszichotrauma és disszociatív kapacitás összefüggésének vizsgálata syncopés betegek körében Dávid Tamás, G. Tóth Kinga, Nagy Kálmán, Rónaszéki Aladár Péterfy S. u. Kórház, Kardiológiai Osztály, Budapest
Pszichotrauma és disszociatív kapacitás összefüggésének vizsgálata syncopés betegek körében Dávid Tamás, G. Tóth Kinga, Nagy Kálmán, Rónaszéki Aladár Péterfy S. u. Kórház, Kardiológiai Osztály, Budapest
A (human)genetika alapja
genetika alapja") A (human)genetika alapja Genom diagnosztika - születés elött - tünetek megjelenése elött - hordozó diagnosztika Prenatalis genetikai diagnosztika indikációi emelkedett valószinüség egy gén betegségre egyik
A (human)genetika alapja Genom diagnosztika - születés elött - tünetek megjelenése elött - hordozó diagnosztika Prenatalis genetikai diagnosztika indikációi emelkedett valószinüség egy gén betegségre egyik
Példák a független öröklődésre
 GENETIKAI PROBLÉMÁK Példák a független öröklődésre Az amelogenesis imperfecta egy, a fogzománc gyengeségével és elszíneződésével járó öröklődő betegség, a 4-es kromoszómán lévő enam gén recesszív mutációja
GENETIKAI PROBLÉMÁK Példák a független öröklődésre Az amelogenesis imperfecta egy, a fogzománc gyengeségével és elszíneződésével járó öröklődő betegség, a 4-es kromoszómán lévő enam gén recesszív mutációja
11. Dr. House. Biokémiai és sejtbiológiai módszerek alkalmazása az orvoslásban
 11. Dr. House. Biokémiai és sejtbiológiai módszerek alkalmazása az orvoslásban HIV fertőzés kimutatása (fiktív) esettanulmány 35 éves nő, HIV fertőzöttség gyanúja. Két partner az elmúlt időszakban. Fertőzött-e
11. Dr. House. Biokémiai és sejtbiológiai módszerek alkalmazása az orvoslásban HIV fertőzés kimutatása (fiktív) esettanulmány 35 éves nő, HIV fertőzöttség gyanúja. Két partner az elmúlt időszakban. Fertőzött-e
Figyelemhiány/Hiperaktivitás Zavar - ADHD TÁJÉKOZTATÓ FÜZET. ADHD-s gyermekek családjai részére
 Figyelemhiány/Hiperaktivitás Zavar - ADHD TÁJÉKOZTATÓ FÜZET ADHD-s gyermekek családjai részére KEZELÉSI TÁJÉKOZTATÓ FÜZET Ezt a tájékoztató füzetet azért készítettük, hogy segítsünk a FIGYELEMHIÁNY/HIPERAKTIVITÁS
Figyelemhiány/Hiperaktivitás Zavar - ADHD TÁJÉKOZTATÓ FÜZET ADHD-s gyermekek családjai részére KEZELÉSI TÁJÉKOZTATÓ FÜZET Ezt a tájékoztató füzetet azért készítettük, hogy segítsünk a FIGYELEMHIÁNY/HIPERAKTIVITÁS
A gyermekek növekedése és fejlődése
 A gyermekek növekedése és fejlődése Körner Anna Gyermekpszichiáter rezidensképzés, 2006. Alacsonynövés Testmagasság a korspecifikus 3-as percentilis (-2 SD) alatt Lassú növekedés A növekedési sebesség
A gyermekek növekedése és fejlődése Körner Anna Gyermekpszichiáter rezidensképzés, 2006. Alacsonynövés Testmagasság a korspecifikus 3-as percentilis (-2 SD) alatt Lassú növekedés A növekedési sebesség
Biológiai módszerek alkalmazása környezeti hatások okozta terhelések kimutatására
 Szalma Katalin Biológiai módszerek alkalmazása környezeti hatások okozta terhelések kimutatására Témavezető: Dr. Turai István, OSSKI Budapest, 2010. október 4. Az ionizáló sugárzás sejt kölcsönhatása Antone
Szalma Katalin Biológiai módszerek alkalmazása környezeti hatások okozta terhelések kimutatására Témavezető: Dr. Turai István, OSSKI Budapest, 2010. október 4. Az ionizáló sugárzás sejt kölcsönhatása Antone
Többgénes jellegek. 1. Klasszikus (poligénes) mennyiségi jellegek. 2.Szinte minden jelleg több gén irányítása alatt áll
 mennyiségi jellegek. 2.Szinte minden jelleg több gén irányítása alatt áll") Többgénes jellegek Többgénes jellegek 1. 1. Klasszikus (poligénes) mennyiségi jellegek Multifaktoriális jellegek: több gén és a környezet által meghatározott jellegek 2.Szinte minden jelleg több gén irányítása
Többgénes jellegek Többgénes jellegek 1. 1. Klasszikus (poligénes) mennyiségi jellegek Multifaktoriális jellegek: több gén és a környezet által meghatározott jellegek 2.Szinte minden jelleg több gén irányítása
A kromoszómák kialakulása előtt a DNS állomány megkettőződik. A két azonos információ tartalmú DNS egymás mellé rendeződik és egy kromoszómát alkot.
 Kromoszómák, Gének A kromoszóma egy hosszú DNS szakasz, amely a sejt életének bizonyos szakaszában (a sejtosztódás előkészítéseként) tömörödik, így fénymikroszkóppal láthatóvá válik. A kromoszómák két
Kromoszómák, Gének A kromoszóma egy hosszú DNS szakasz, amely a sejt életének bizonyos szakaszában (a sejtosztódás előkészítéseként) tömörödik, így fénymikroszkóppal láthatóvá válik. A kromoszómák két
Legújabb tudományos eredmények a Rett-szindróma terápiás lehetőségeinek kutatásában
 Legújabb tudományos eredmények a Rett-szindróma terápiás lehetőségeinek kutatásában Dr. Varga Orsolya Budapest, 2015. január. 24 A kutatás a TÁMOP-4.2.4.A/2-11/1-2012-0001 Nemzeti Kiválóság Program című
Legújabb tudományos eredmények a Rett-szindróma terápiás lehetőségeinek kutatásában Dr. Varga Orsolya Budapest, 2015. január. 24 A kutatás a TÁMOP-4.2.4.A/2-11/1-2012-0001 Nemzeti Kiválóság Program című
Dr. Nemes Nagy Zsuzsa Szakképzés Karl Landsteiner Karl Landsteiner:
 Az AB0 vércsoport rendszer Dr. Nemes Nagy Zsuzsa Szakképzés 2011 Az AB0 rendszer felfedezése 1901. Karl Landsteiner Landsteiner szabály 1901 Karl Landsteiner: Munkatársai vérmintáit vizsgálva fedezte fel
Az AB0 vércsoport rendszer Dr. Nemes Nagy Zsuzsa Szakképzés 2011 Az AB0 rendszer felfedezése 1901. Karl Landsteiner Landsteiner szabály 1901 Karl Landsteiner: Munkatársai vérmintáit vizsgálva fedezte fel
Genomika. Mutációk (SNP-k) és vizsgálatuk egyszerű módszerekkel. DNS szekvenálási eljárások. DNS ujjlenyomat (VNTR)
 és vizsgálatuk egyszerű módszerekkel. DNS szekvenálási eljárások. DNS ujjlenyomat (VNTR)") Genomika (A genom, génállomány vizsgálata) Mutációk (SNP-k) és vizsgálatuk egyszerű módszerekkel DNS szekvenálási eljárások DNS ujjlenyomat (VNTR) DNS chipek statikus és dinamikus információk vizsgálata
Genomika (A genom, génállomány vizsgálata) Mutációk (SNP-k) és vizsgálatuk egyszerű módszerekkel DNS szekvenálási eljárások DNS ujjlenyomat (VNTR) DNS chipek statikus és dinamikus információk vizsgálata
A World Health Organisation diagnosztikus rendszerébõl BNO-10 (1995) (Az ICD-10, 1992 fordítása)
 (Az ICD-10, 1992 fordítása)") A World Health Organisation diagnosztikus rendszerébõl BNO-10 (1995) (Az ICD-10, 1992 fordítása) F84 - Pervasiv fejlõdési zavarok A zavaroknak egy olyan alcsoportja, melyben a reciprok szociális interakciók,
A World Health Organisation diagnosztikus rendszerébõl BNO-10 (1995) (Az ICD-10, 1992 fordítása) F84 - Pervasiv fejlõdési zavarok A zavaroknak egy olyan alcsoportja, melyben a reciprok szociális interakciók,
A tremor elektrofiziológiai vizsgálata mozgászavarral járó kórképekben. Doktori tézisek. Dr. Farkas Zsuzsanna
 A tremor elektrofiziológiai vizsgálata mozgászavarral járó kórképekben Doktori tézisek Dr. Farkas Zsuzsanna Semmelweis Egyetem Szentágothai János Idegtudományi Doktori Iskola Témavezető: Dr. Kamondi Anita
A tremor elektrofiziológiai vizsgálata mozgászavarral járó kórképekben Doktori tézisek Dr. Farkas Zsuzsanna Semmelweis Egyetem Szentágothai János Idegtudományi Doktori Iskola Témavezető: Dr. Kamondi Anita
Esettanulmányok. Dr. Toldi József OVSZ Szegedi Regionális Vérellátó Központ
 Esettanulmányok Dr. Toldi József OVSZ Szegedi Regionális Vérellátó Központ Esetismertetések: ABO vércsoport meghatározással kapcsolatos nehézségek és megoldások RhD bizonytalanság kivizsgálása Antitest-azonosítással
Esettanulmányok Dr. Toldi József OVSZ Szegedi Regionális Vérellátó Központ Esetismertetések: ABO vércsoport meghatározással kapcsolatos nehézségek és megoldások RhD bizonytalanság kivizsgálása Antitest-azonosítással
MAGYOT évi Tudományos Szimpóziuma Május 5-6, Budapest
 MAGYOT 2017. évi Tudományos Szimpóziuma Május 5-6, Budapest A petefészekrákok kezelésében nem régen került bevezetésre egy újabb fenntartó kezelés BRCA mutációt hordozó (szomatikus vagy germinális) magas
MAGYOT 2017. évi Tudományos Szimpóziuma Május 5-6, Budapest A petefészekrákok kezelésében nem régen került bevezetésre egy újabb fenntartó kezelés BRCA mutációt hordozó (szomatikus vagy germinális) magas
ÁLLATOK KLINIKAI VIZSGÁLATAI
 ÁLLATOK KLINIKAI VIZSGÁLATAI ---------------------------------------------------------------------------------------------------- Állatokon végzett tanulmányok A CV247 két kutatásban képezte vizsgálat
ÁLLATOK KLINIKAI VIZSGÁLATAI ---------------------------------------------------------------------------------------------------- Állatokon végzett tanulmányok A CV247 két kutatásban képezte vizsgálat
mintasepcifikus mikrokapilláris elektroforézis Lab-on-Chip elektroforézis / elektrokinetikus elven DNS, RNS, mirns 12, fehérje 10, sejtes minta 6
 Agilent 2100 Bioanalyzer mikrokapilláris gélelektroforézis rendszer G2943CA 2100 Bioanalyzer system forgalmazó: Kromat Kft. 1112 Budapest Péterhegyi u. 98. t:36 (1) 248-2110 www.kromat.hu bio@kromat.hu
Agilent 2100 Bioanalyzer mikrokapilláris gélelektroforézis rendszer G2943CA 2100 Bioanalyzer system forgalmazó: Kromat Kft. 1112 Budapest Péterhegyi u. 98. t:36 (1) 248-2110 www.kromat.hu bio@kromat.hu
Sürgős ellátás kora gyermekkorban multifaktoriálisszempontok szerint. Scheuring Noémi Heim Pál Gyermekkórház, Budapest
 Sürgős ellátás kora gyermekkorban multifaktoriálisszempontok szerint Scheuring Noémi Heim Pál Gyermekkórház, Budapest Amikor a sürgős ellátás szükségessége felmerül Probléma lép fel Gyerek: A tünet megjelenésének
Sürgős ellátás kora gyermekkorban multifaktoriálisszempontok szerint Scheuring Noémi Heim Pál Gyermekkórház, Budapest Amikor a sürgős ellátás szükségessége felmerül Probléma lép fel Gyerek: A tünet megjelenésének
III./3.2.3.3. Egyes dystonia szindrómák. III./3.2.3.3.1. Blepharospasmus
 III./3.2.3.3. Egyes dystonia szindrómák III./3.2.3.3.1. Blepharospasmus A blepharospasmus jellemzője a szemrést záró izomzat (m. orbicularis oculi) tartós vagy intermittáló, kétoldali akaratlan kontrakciója.
III./3.2.3.3. Egyes dystonia szindrómák III./3.2.3.3.1. Blepharospasmus A blepharospasmus jellemzője a szemrést záró izomzat (m. orbicularis oculi) tartós vagy intermittáló, kétoldali akaratlan kontrakciója.
ADATBÁNYÁSZAT I. ÉS OMICS
 Az élettudományi-klinikai felsőoktatás gyakorlatorientált és hallgatóbarát korszerűsítése a vidéki képzőhelyek nemzetközi versenyképességének erősítésére TÁMOP-4.1.1.C-13/1/KONV-2014-0001 ADATBÁNYÁSZAT
Az élettudományi-klinikai felsőoktatás gyakorlatorientált és hallgatóbarát korszerűsítése a vidéki képzőhelyek nemzetközi versenyképességének erősítésére TÁMOP-4.1.1.C-13/1/KONV-2014-0001 ADATBÁNYÁSZAT
Tüdő adenocarcinomásbetegek agyi áttéteiben jelenlévő immunsejtek, valamint a PD-L1 és PD-1 fehérjék túlélésre gyakorolt hatása
 Tüdő adenocarcinomásbetegek agyi áttéteiben jelenlévő immunsejtek, valamint a és PD-1 fehérjék túlélésre gyakorolt hatása Téglási Vanda, MoldvayJudit, Fábián Katalin, Csala Irén, PipekOrsolya, Bagó Attila,
Tüdő adenocarcinomásbetegek agyi áttéteiben jelenlévő immunsejtek, valamint a és PD-1 fehérjék túlélésre gyakorolt hatása Téglási Vanda, MoldvayJudit, Fábián Katalin, Csala Irén, PipekOrsolya, Bagó Attila,
Genomikai Medicina és Ritka Betegségek Intézete Semmelweis Egyetem
 Tisztelt Hölgyem, Tisztelt Uram! Örömmel jelentjük be Önöknek, hogy a Genomikai Medicina és Ritka Betegségek Intézetének egyik új projektje azon betegségek genetikai hátterének feltérképezésére irányul,
Tisztelt Hölgyem, Tisztelt Uram! Örömmel jelentjük be Önöknek, hogy a Genomikai Medicina és Ritka Betegségek Intézetének egyik új projektje azon betegségek genetikai hátterének feltérképezésére irányul,
Alkohollal kapcsolatos zavarok. Az alkoholbetegség. Általános jellegzetességek
 Alkohollal kapcsolatos zavarok Az alkoholbetegség Az alkoholisták mértéktelen ivók, alkoholfüggőségük olyan szintet ér el, hogy észrevehető mentális zavarokat okoz, károsítja test-lelki egészségüket, interperszonális
Alkohollal kapcsolatos zavarok Az alkoholbetegség Az alkoholisták mértéktelen ivók, alkoholfüggőségük olyan szintet ér el, hogy észrevehető mentális zavarokat okoz, károsítja test-lelki egészségüket, interperszonális
A termesztett búza diploid őseinek molekuláris citogenetikai elemzése: pachytén- és fiber-fish.
 OTKA K67808 zárójelentés 2012. A termesztett búza diploid őseinek molekuláris citogenetikai elemzése: pachytén- és fiber-fish. A fluoreszcens in situ hibridizáció (FISH) olyan technikai fejlettséget ért
OTKA K67808 zárójelentés 2012. A termesztett búza diploid őseinek molekuláris citogenetikai elemzése: pachytén- és fiber-fish. A fluoreszcens in situ hibridizáció (FISH) olyan technikai fejlettséget ért
Rácz Olivér, Ništiar Ferenc, Hubka Beáta, Miskolci Egyetem, Egészségügyi Kar 2010
 Kromoszóma eltérések Rácz Olivér, Ništiar Ferenc, Hubka Beáta, Miskolci Egyetem, Egészségügyi Kar 2010 20.3.2010 genmisk6.ppt 1 Alapfogalmak ismétlése Csak a sejtosztódás közben láthatóak (de természetesen
Kromoszóma eltérések Rácz Olivér, Ništiar Ferenc, Hubka Beáta, Miskolci Egyetem, Egészségügyi Kar 2010 20.3.2010 genmisk6.ppt 1 Alapfogalmak ismétlése Csak a sejtosztódás közben láthatóak (de természetesen
Embriószelekció PGD-vel genetikai terheltség esetén. Kónya Márton Istenhegyi Géndiagnosztika
 Embriószelekció PGD-vel genetikai terheltség esetén Kónya Márton Istenhegyi Géndiagnosztika A praeimplantatiós genetikai diagnosztika (PGD) a praenatalis diagnosztika legkorábbi formája, a beágyazódás
Embriószelekció PGD-vel genetikai terheltség esetén Kónya Márton Istenhegyi Géndiagnosztika A praeimplantatiós genetikai diagnosztika (PGD) a praenatalis diagnosztika legkorábbi formája, a beágyazódás
Lymphoma sejtvonalak és gyerekkori leukémia (ALL) sejtek mikro RNS (mir) profiljának vizsgálata
 sejtek mikro RNS (mir) profiljának vizsgálata") Lymphoma sejtvonalak és gyerekkori leukémia (ALL) sejtek mikro RNS (mir) profiljának vizsgálata Dr. Nemes Karolina, Márk Ágnes, Dr. Hajdu Melinda, Csorba Gézáné, Dr. Kopper László, Dr. Csóka Monika, Dr.
Lymphoma sejtvonalak és gyerekkori leukémia (ALL) sejtek mikro RNS (mir) profiljának vizsgálata Dr. Nemes Karolina, Márk Ágnes, Dr. Hajdu Melinda, Csorba Gézáné, Dr. Kopper László, Dr. Csóka Monika, Dr.
NÖVÉNYGENETIKA. Az Agrármérnöki MSc szak tananyagfejlesztése TÁMOP /1/A
 NÖVÉNYGENETIKA Az Agrármérnöki MSc szak tananyagfejlesztése TÁMOP-4.1.2-08/1/A-2009-0010 A citológia és a genetika társtudománya Citogenetika A kromoszómák eredetét, szerkezetét, genetikai funkcióját,
NÖVÉNYGENETIKA Az Agrármérnöki MSc szak tananyagfejlesztése TÁMOP-4.1.2-08/1/A-2009-0010 A citológia és a genetika társtudománya Citogenetika A kromoszómák eredetét, szerkezetét, genetikai funkcióját,
Humán genom variációk single nucleotide polymorphism (SNP)
") Humán genom variációk single nucleotide polymorphism (SNP) A genom ~ 97 %-a két különböző egyedben teljesen azonos ~ 1% különbség: SNP miatt ~2% különbség: kópiaszámbeli eltérés, deléciók miatt 11-12 millió
Humán genom variációk single nucleotide polymorphism (SNP) A genom ~ 97 %-a két különböző egyedben teljesen azonos ~ 1% különbség: SNP miatt ~2% különbség: kópiaszámbeli eltérés, deléciók miatt 11-12 millió
Az agy betegségeinek molekuláris biológiája. 1. Prion betegség 2. Trinukleotid ripít betegségek 3. ALS 4. Parkinson kór 5.
 Az agy betegségeinek molekuláris biológiája 1. Prion betegség 2. Trinukleotid ripít betegségek 3. ALS 4. Parkinson kór 5. Alzheimer kór 28 Prion betegség A prion betegség fertőző formáját nem egy genetikai
Az agy betegségeinek molekuláris biológiája 1. Prion betegség 2. Trinukleotid ripít betegségek 3. ALS 4. Parkinson kór 5. Alzheimer kór 28 Prion betegség A prion betegség fertőző formáját nem egy genetikai
A pszichomotoros fejlődés zavarainak felismerése és ellátása
 A pszichomotoros fejlődés zavarainak felismerése és ellátása Dr. Gallai Mária gyermekpszichiáter Fejlődési zavar Fejlődési zavar gyanúja: megkésett eltérő disszociált fejlődés Fejlődési részterületek:
A pszichomotoros fejlődés zavarainak felismerése és ellátása Dr. Gallai Mária gyermekpszichiáter Fejlődési zavar Fejlődési zavar gyanúja: megkésett eltérő disszociált fejlődés Fejlődési részterületek:
67. Pathologus Kongresszus
 A kemoirradiáció okozta oncocytás átalakulás szövettani, immunhisztokémiai, ultrastruktúrális jellemzői és lehetséges prognosztikus jelentősége rectum adenocarcinomákban 67. Pathologus Kongresszus Bogner
A kemoirradiáció okozta oncocytás átalakulás szövettani, immunhisztokémiai, ultrastruktúrális jellemzői és lehetséges prognosztikus jelentősége rectum adenocarcinomákban 67. Pathologus Kongresszus Bogner
A keringı tumor markerek klinikai alkalmazásának aktuális kérdései és irányelvei
 A keringı tumor markerek klinikai alkalmazásának aktuális kérdései és irányelvei A TM vizsgálatok alapkérdései A vizsgálatok célja, információértéke? Az alkalmazás területei? Hogyan válasszuk ki az alkalmazott
A keringı tumor markerek klinikai alkalmazásának aktuális kérdései és irányelvei A TM vizsgálatok alapkérdései A vizsgálatok célja, információértéke? Az alkalmazás területei? Hogyan válasszuk ki az alkalmazott
A bioinformatika gyökerei
 A bioinformatika gyökerei 1944: Avery a transforming principle a DNS 1952: Hershey és Chase perdöntő bizonyíték: a bakteriofágok szaporodásakor csak a DNS jut be a sejtbe 1953: Watson és Crick a DNS szerkezete
A bioinformatika gyökerei 1944: Avery a transforming principle a DNS 1952: Hershey és Chase perdöntő bizonyíték: a bakteriofágok szaporodásakor csak a DNS jut be a sejtbe 1953: Watson és Crick a DNS szerkezete
Epilepszia és görcsállapotok gyermekkorban. Fogarasi András. Bethesda Gyermekkórház, Budapest. Gyermekgyógyászati kötelező szinten tartó tanfolyam
 Epilepszia és görcsállapotok gyermekkorban Fogarasi András Bethesda Gyermekkórház, Budapest Gyermekgyógyászati kötelező szinten tartó tanfolyam Budapest, 2013. november 22. fog.andras@gmail.com Tel: 4222-875
Epilepszia és görcsállapotok gyermekkorban Fogarasi András Bethesda Gyermekkórház, Budapest Gyermekgyógyászati kötelező szinten tartó tanfolyam Budapest, 2013. november 22. fog.andras@gmail.com Tel: 4222-875
Rh VÉRCSOPORT RENDSZER GENETIKÁJA. Rh ANTIGÉNEK ÉS ANTITESTEK. EGYÉB VÉRCSOPORTRENDSZEREK
 Rh VÉRCSOPORT RENDSZER GENETIKÁJA. Rh ANTIGÉNEK ÉS ANTITESTEK. EGYÉB VÉRCSOPORTRENDSZEREK HISTORY Antitestet találtak egy koraszülött gyermek anyjának szérumában; ez lenne felelős a gyermek haláláért?
Rh VÉRCSOPORT RENDSZER GENETIKÁJA. Rh ANTIGÉNEK ÉS ANTITESTEK. EGYÉB VÉRCSOPORTRENDSZEREK HISTORY Antitestet találtak egy koraszülött gyermek anyjának szérumában; ez lenne felelős a gyermek haláláért?
Az ember összes kromoszómája 23 párt alkot. A 23. pár határozza meg a nemünket. Ha 2 db X kromoszómánk van ezen a helyen, akkor nők, ha 1db X és 1db
 Testünk minden sejtjében megtalálhatók a kromoszómák, melyek a tulajdonságok átörökítését végzik. A testi sejtekben 2 x 23 = 46 db kromoszóma van. Az egyik sorozat apánktól, a másik anyánktól származik.
Testünk minden sejtjében megtalálhatók a kromoszómák, melyek a tulajdonságok átörökítését végzik. A testi sejtekben 2 x 23 = 46 db kromoszóma van. Az egyik sorozat apánktól, a másik anyánktól származik.
Markov modellek 2015.03.19.
 Markov modellek 2015.03.19. Markov-láncok Markov-tulajdonság: egy folyamat korábbi állapotai a későbbiekre csak a jelen állapoton keresztül gyakorolnak befolyást. Semmi, ami a múltban történt, nem ad előrejelzést
Markov modellek 2015.03.19. Markov-láncok Markov-tulajdonság: egy folyamat korábbi állapotai a későbbiekre csak a jelen állapoton keresztül gyakorolnak befolyást. Semmi, ami a múltban történt, nem ad előrejelzést
Ha nem akarsz mellé-nyúl-ni, használj Nobivac Myxo-RHD-t! MSDay-MOM park, 2013.02.21. dr. Schweickhardt Eszter
 Ha nem akarsz mellé-nyúl-ni, használj Nobivac Myxo-RHD-t! MSDay-MOM park, 2013.02.21. dr. Schweickhardt Eszter Áttekintés Miért éppen a nyuszik? Védekezés módja Vakcina jellemzői Vakcina működése Ráfertőzési
Ha nem akarsz mellé-nyúl-ni, használj Nobivac Myxo-RHD-t! MSDay-MOM park, 2013.02.21. dr. Schweickhardt Eszter Áttekintés Miért éppen a nyuszik? Védekezés módja Vakcina jellemzői Vakcina működése Ráfertőzési
Az előadás címe: A nyelvi zavarok korai felismerése a pszichomotoros fejlődéssel összefüggésben Egy szakdolgozati kutatás eredményeinek bemutatása
 Az előadás címe: A nyelvi zavarok korai felismerése a pszichomotoros fejlődéssel összefüggésben Egy szakdolgozati kutatás eredményeinek bemutatása Készítette: Szabó Ágnes logopédus hallgató; ELTE Bárczi
Az előadás címe: A nyelvi zavarok korai felismerése a pszichomotoros fejlődéssel összefüggésben Egy szakdolgozati kutatás eredményeinek bemutatása Készítette: Szabó Ágnes logopédus hallgató; ELTE Bárczi
PETEFÉSZEK ELÉGTELENSÉG
 PETEFÉSZEK ELÉGTELENSÉG Dr. Beke Artúr Semmelweis Egyetem I. Szülészeti és Nőgyógyászati Klinika Bevezetés POI (Primary Ovarian Insufficiency - primer petefészekelégtelenség) POF (Premature Ovarian Failure
PETEFÉSZEK ELÉGTELENSÉG Dr. Beke Artúr Semmelweis Egyetem I. Szülészeti és Nőgyógyászati Klinika Bevezetés POI (Primary Ovarian Insufficiency - primer petefészekelégtelenség) POF (Premature Ovarian Failure
Diagnosztikai célú molekuláris biológiai vizsgálatok
 Diagnosztikai célú molekuláris biológiai vizsgálatok Dr. Patócs Attila, PhD MTA-SE Molekuláris Medicina Kutatócsoport, Semmelweis Egyetem II. sz. Belgyógyászati Klinika Laboratóriumi Medicina Intézet Genetikai
Diagnosztikai célú molekuláris biológiai vizsgálatok Dr. Patócs Attila, PhD MTA-SE Molekuláris Medicina Kutatócsoport, Semmelweis Egyetem II. sz. Belgyógyászati Klinika Laboratóriumi Medicina Intézet Genetikai
ELŐADÁS VÁZLAT. Balázs Judit
 ELŐADÁS VÁZLAT GYERMEKKORBAN KEZDŐDŐ FELNŐTT PSZICHIÁTRIAI KÓRKÉPEK: AUTIZMUS, ADHD, TIC-ZAVAR Balázs Judit 2012. november 15-17. SEMMELWEIS EGYETEM, KÖTELEZŐ SZINTEN TARTÓ TANFOLYAM AUTIZMUS FOGALMI SOKASÁG
ELŐADÁS VÁZLAT GYERMEKKORBAN KEZDŐDŐ FELNŐTT PSZICHIÁTRIAI KÓRKÉPEK: AUTIZMUS, ADHD, TIC-ZAVAR Balázs Judit 2012. november 15-17. SEMMELWEIS EGYETEM, KÖTELEZŐ SZINTEN TARTÓ TANFOLYAM AUTIZMUS FOGALMI SOKASÁG
Anamnézis - Kórelőzmény
 Anamnézis - Kórelőzmény 2016 Ifél év Prof. Dr. Szabó András egyetemi tanár Semmelweis Egyetem II. sz. Gyermekklinika A beteg kikérdezése a betegség előzményinek,okainak feltárása céljából A jó anamnézis
Anamnézis - Kórelőzmény 2016 Ifél év Prof. Dr. Szabó András egyetemi tanár Semmelweis Egyetem II. sz. Gyermekklinika A beteg kikérdezése a betegség előzményinek,okainak feltárása céljából A jó anamnézis
2. SZ. SZAKMAI ÖSSZEFOGLALÓ PIR 2
 Az Országos Onkológiai Intézet Norvég Finanszírozási mechanizmus keretében elnyert Kutatás Fejlesztési Pályázata Közös stratégia kifejlesztése molekuláris módszerek alkalmazásával a rák kezelésére Magyarországon
Az Országos Onkológiai Intézet Norvég Finanszírozási mechanizmus keretében elnyert Kutatás Fejlesztési Pályázata Közös stratégia kifejlesztése molekuláris módszerek alkalmazásával a rák kezelésére Magyarországon
2354-06 Nőgyógyászati citodiagnosztika követelménymodul szóbeli vizsgafeladatai
 1. feladat A laboratóriumba vendég érkezik. Tájékoztassa a rákmegelőzés lehetőségeiről! Ismertesse a citológiai előszűrő vizsgálatok lényegét! - a primer és szekunder prevenció fogalma, eszközei - a citológia
1. feladat A laboratóriumba vendég érkezik. Tájékoztassa a rákmegelőzés lehetőségeiről! Ismertesse a citológiai előszűrő vizsgálatok lényegét! - a primer és szekunder prevenció fogalma, eszközei - a citológia
Áramlási citometria, sejtszeparációs technikák. Immunológiai és Biotechnológiai Intézet ÁOK, PTE
 Áramlási citometria, sejtszeparációs technikák Immunológiai és Biotechnológiai Intézet ÁOK, PTE Az áramlási citometria elve Az áramlási citometria ( Flow cytometria ) sejtek gyors, multiparaméteres vizsgálatára
Áramlási citometria, sejtszeparációs technikák Immunológiai és Biotechnológiai Intézet ÁOK, PTE Az áramlási citometria elve Az áramlási citometria ( Flow cytometria ) sejtek gyors, multiparaméteres vizsgálatára
Tóth Erika Sebészeti és Molekuláris Patológia Osztály Országos Onkológiai Intézet. Frank Diagnosztika Szimpózium, DAKO workshop 2012.
 Tóth Erika Sebészeti és Molekuláris Patológia Osztály Országos Onkológiai Intézet Frank Diagnosztika Szimpózium, DAKO workshop 2012.december 7 Follicularis hám eredetű daganatok Jól differenciált tumorok
Tóth Erika Sebészeti és Molekuláris Patológia Osztály Országos Onkológiai Intézet Frank Diagnosztika Szimpózium, DAKO workshop 2012.december 7 Follicularis hám eredetű daganatok Jól differenciált tumorok
Genetikai vizsgálatok
 Genetikai vizsgálatok Családvizsgálatok Bottom-up Top-down Ikertanulmányok Adoptációs vizsgálatok Molekuláris genetikai vizsgálatok 1 Depresszió-Családvizsgálatok Bottom-up tanulmányok (MDD-s gyermek proband
Genetikai vizsgálatok Családvizsgálatok Bottom-up Top-down Ikertanulmányok Adoptációs vizsgálatok Molekuláris genetikai vizsgálatok 1 Depresszió-Családvizsgálatok Bottom-up tanulmányok (MDD-s gyermek proband
A Hardy-Weinberg egyensúly. 2. gyakorlat
 A Hardy-Weinberg egyensúly 2. gyakorlat A Hardy-Weinberg egyensúly feltételei: nincs szelekció nincs migráció nagy populációméret (nincs sodródás) nincs mutáció pánmixis van allélgyakoriság azonos hímekben
A Hardy-Weinberg egyensúly 2. gyakorlat A Hardy-Weinberg egyensúly feltételei: nincs szelekció nincs migráció nagy populációméret (nincs sodródás) nincs mutáció pánmixis van allélgyakoriság azonos hímekben
A DEMOGRÁFIÁI ÉS A SZOCIOLÓGIAI ÉLETRAJZ EGYESÍTÉSE A NŐI ÉLETÜT V IZSG ÁLATA ALAPJÁN DR. M O LNÁR LÁSZLÓ
 A DEMOGRÁFIÁI ÉS A SZOCIOLÓGIAI ÉLETRAJZ EGYESÍTÉSE A NŐI ÉLETÜT V IZSG ÁLATA ALAPJÁN DR. M O LNÁR LÁSZLÓ A gazdaságilag aktív nő életútjának, életciklusainak kutatását bemutató tanulmányomban (Molnár,
A DEMOGRÁFIÁI ÉS A SZOCIOLÓGIAI ÉLETRAJZ EGYESÍTÉSE A NŐI ÉLETÜT V IZSG ÁLATA ALAPJÁN DR. M O LNÁR LÁSZLÓ A gazdaságilag aktív nő életútjának, életciklusainak kutatását bemutató tanulmányomban (Molnár,
Mentális retardáció. PDF created with pdffactory Pro trial version
 Mentális retardáció 1 Definíció: IQ
Mentális retardáció 1 Definíció: IQ
A stroke betegek rehabilitációja során felmerülő nehézségek elemzése ápolói szemszögből
 ORFMMT XXXII. Vándorgyűlése Miskolc, 2013.08.29-2013.08.31 A stroke betegek rehabilitációja során felmerülő nehézségek elemzése ápolói szemszögből T E N K S Z A B I N A D R. U R B Á N E D I N A Kutatás
ORFMMT XXXII. Vándorgyűlése Miskolc, 2013.08.29-2013.08.31 A stroke betegek rehabilitációja során felmerülő nehézségek elemzése ápolói szemszögből T E N K S Z A B I N A D R. U R B Á N E D I N A Kutatás
PrenaTest Újgenerációs szekvenálást és z-score
 PrenaTest Újgenerációs szekvenálást és z-score számítást alkalmazó, nem-invazív prenatális molekuláris genetikai teszt a magzati 21-es triszómia észlelésére, anyai vérből végzett DNS izolálást követően
PrenaTest Újgenerációs szekvenálást és z-score számítást alkalmazó, nem-invazív prenatális molekuláris genetikai teszt a magzati 21-es triszómia észlelésére, anyai vérből végzett DNS izolálást követően
Minden leendő szülő számára a legfontosabb, hogy születendő gyermeke egészséges legyen. A súlyosan beteg gyermek komoly terheket ró a családra.
 Egészséges magzat, biztonságos jövő Minden leendő szülő számára a legfontosabb, hogy születendő gyermeke egészséges legyen. A súlyosan beteg gyermek komoly terheket ró a családra. A veleszületett fejlődési
Egészséges magzat, biztonságos jövő Minden leendő szülő számára a legfontosabb, hogy születendő gyermeke egészséges legyen. A súlyosan beteg gyermek komoly terheket ró a családra. A veleszületett fejlődési
Tesztkérdések. 1 A nem fertőző betegségek halálozási aránya az európai régióban: a) 77% b) 68% c) 86%
 77% b) 68% c) 86%") Tesztkérdések 1 A nem fertőző betegségek halálozási aránya az európai régióban: a) 77% b) 68% c) 86% 2. Az étkezés és az életmód megváltoztatásával milyen arányban előzhetők meg a szívérrendszeri betegségek?
Tesztkérdések 1 A nem fertőző betegségek halálozási aránya az európai régióban: a) 77% b) 68% c) 86% 2. Az étkezés és az életmód megváltoztatásával milyen arányban előzhetők meg a szívérrendszeri betegségek?
Vezetői összefoglaló a Veleszületett Rendellenességek Országos Nyilvántartása (VRONY) évi adataiból készült jelentésről
 évi adataiból készült jelentésről") Vezetői összefoglaló a Veleszületett Rendellenességek Országos Nyilvántartása (VRONY) 2010. évi adataiból készült jelentésről Veleszületett Rendellenességek Országos Felügyeleti Osztálya Magyarországon
Vezetői összefoglaló a Veleszületett Rendellenességek Országos Nyilvántartása (VRONY) 2010. évi adataiból készült jelentésről Veleszületett Rendellenességek Országos Felügyeleti Osztálya Magyarországon
Mit tud a genetika. Génterápiás lehetőségek MPS-ben. Dr. Varga Norbert
 Mit tud a genetika Génterápiás lehetőségek MPS-ben Dr. Varga Norbert Oki terápia Terápiás lehetőségek MPS-ben A kiváltó okot gyógyítja meg ERT Enzimpótló kezelés Őssejt transzplantáció Genetikai beavatkozások
Mit tud a genetika Génterápiás lehetőségek MPS-ben Dr. Varga Norbert Oki terápia Terápiás lehetőségek MPS-ben A kiváltó okot gyógyítja meg ERT Enzimpótló kezelés Őssejt transzplantáció Genetikai beavatkozások
Regulációs zavarok kutatása az Egészséges utódokért program keretében
 Regulációs zavarok kutatása az Egészséges utódokért program keretében Scheuring N.(1); Danis I.(2); Németh T.(3); Papp E.(1); Czinner Antal Prof.(1) Heim Pál Gyermekkórház, Budapest, Belgyógyászat (1);
Regulációs zavarok kutatása az Egészséges utódokért program keretében Scheuring N.(1); Danis I.(2); Németh T.(3); Papp E.(1); Czinner Antal Prof.(1) Heim Pál Gyermekkórház, Budapest, Belgyógyászat (1);
NOAC-kezelés pitvarfibrillációban. Thrombolysis, thrombectomia és kombinációja. Az ischaemiás kórképek szekunder prevenciója. A TIA új, szöveti alapú
 NOAC-kezelés pitvarfibrillációban. Thrombolysis, thrombectomia és kombinációja. Az ischaemiás kórképek szekunder prevenciója. A TIA új, szöveti alapú meghatározása. (Megj.: a felsorolt esetekben meghatározó
NOAC-kezelés pitvarfibrillációban. Thrombolysis, thrombectomia és kombinációja. Az ischaemiás kórképek szekunder prevenciója. A TIA új, szöveti alapú meghatározása. (Megj.: a felsorolt esetekben meghatározó
Súlyos infekciók differenciálása a rendelőben. Dr. Fekete Ferenc Heim Pál Gyermekkórház Madarász utcai Gyermekkórháza
 Súlyos infekciók differenciálása a rendelőben Dr. Fekete Ferenc Heim Pál Gyermekkórház Madarász utcai Gyermekkórháza Miért probléma a lázas gyermek a rendelőben? nem beteg - súlyos beteg otthon ellátható
Súlyos infekciók differenciálása a rendelőben Dr. Fekete Ferenc Heim Pál Gyermekkórház Madarász utcai Gyermekkórháza Miért probléma a lázas gyermek a rendelőben? nem beteg - súlyos beteg otthon ellátható
TUMORSEJTEK FENOTÍPUS-VÁLTOZÁSA TUMOR-SZTRÓMA SEJTFÚZIÓ HATÁSÁRA. Dr. Kurgyis Zsuzsanna
 TUMORSEJTEK FENOTÍPUS-VÁLTOZÁSA TUMOR-SZTRÓMA SEJTFÚZIÓ HATÁSÁRA PhD tézis Dr. Kurgyis Zsuzsanna Bőrgyógyászati és Allergológiai Klinika Szegedi Tudományegyetem Szeged 2017 2 TUMORSEJTEK FENOTÍPUS-VÁLTOZÁSA
TUMORSEJTEK FENOTÍPUS-VÁLTOZÁSA TUMOR-SZTRÓMA SEJTFÚZIÓ HATÁSÁRA PhD tézis Dr. Kurgyis Zsuzsanna Bőrgyógyászati és Allergológiai Klinika Szegedi Tudományegyetem Szeged 2017 2 TUMORSEJTEK FENOTÍPUS-VÁLTOZÁSA
Tájékoztató a Down szűrésről Első trimeszteri KOMBINÁLT TESZT
 Tájékoztató a Down szűrésről Első trimeszteri KOMBINÁLT TESZT A terhességek kb. 1%-ában az újszülött teljesen egészséges szülőktől súlyos szellemi vagy testi fogyatékkal születik. A veleszületett értelmi
Tájékoztató a Down szűrésről Első trimeszteri KOMBINÁLT TESZT A terhességek kb. 1%-ában az újszülött teljesen egészséges szülőktől súlyos szellemi vagy testi fogyatékkal születik. A veleszületett értelmi
A korai magömlés diagnózisa és terápiája (ISSM 2014-es ajánlása alapján) Dr. Rosta Gábor Soproni Gyógyközpont
 Dr. Rosta Gábor Soproni Gyógyközpont") A korai magömlés diagnózisa és terápiája (ISSM 2014-es ajánlása alapján) Dr. Rosta Gábor Soproni Gyógyközpont ISSM definíció- 2014 Szexuális aktivitás során az ejakuláció állandóan vagy visszatérően bekövetkezik
A korai magömlés diagnózisa és terápiája (ISSM 2014-es ajánlása alapján) Dr. Rosta Gábor Soproni Gyógyközpont ISSM definíció- 2014 Szexuális aktivitás során az ejakuláció állandóan vagy visszatérően bekövetkezik
CYP2C19 polimorfizmusok szerepe a clopidogrel rezisztencia vizsgálatában iszkémiás stroke-on átesett betegekben
 CYP2C19 polimorfizmusok szerepe a clopidogrel rezisztencia vizsgálatában iszkémiás stroke-on átesett betegekben Bádogos Ágnes DE-ÁOK V. évfolyam Témavezető: Dr. Bagoly Zsuzsa Debreceni Egyetem, Általános
CYP2C19 polimorfizmusok szerepe a clopidogrel rezisztencia vizsgálatában iszkémiás stroke-on átesett betegekben Bádogos Ágnes DE-ÁOK V. évfolyam Témavezető: Dr. Bagoly Zsuzsa Debreceni Egyetem, Általános
AZ EGÉSZSÉGI ÁLLAPOT EGYENLŐTLENSÉGEI
 6. AZ EGÉSZSÉGI ÁLLAPOT EGYENLŐTLENSÉGEI Kovács Katalin FŐBB MEGÁLLAPÍTÁSOK 2009-ben jelentős különbségek mutatkoznak a különböző társadalmi csoportok egészségi állapotában. Az egészségi állapot szoros
6. AZ EGÉSZSÉGI ÁLLAPOT EGYENLŐTLENSÉGEI Kovács Katalin FŐBB MEGÁLLAPÍTÁSOK 2009-ben jelentős különbségek mutatkoznak a különböző társadalmi csoportok egészségi állapotában. Az egészségi állapot szoros
Spondyloarthritisekhez társuló csontvesztés megelőzésének és kezelésének korszerű szemlélete
 Spondyloarthritisekhez társuló csontvesztés megelőzésének és kezelésének korszerű szemlélete Szántó Sándor DE OEC, Reumatológiai Tanszék 2014.02.05. Szeminárium Szisztémás OP SPA-ban Csigolyatörés SPA-s
Spondyloarthritisekhez társuló csontvesztés megelőzésének és kezelésének korszerű szemlélete Szántó Sándor DE OEC, Reumatológiai Tanszék 2014.02.05. Szeminárium Szisztémás OP SPA-ban Csigolyatörés SPA-s
Johann Gregor Mendel Az olmüci (Olomouc) és bécsi egyetem diákja Brünni ágostonrendi apát (nem szovjet tudós) Tudatos és nagyon alapos kutat
 és bécsi egyetem diákja Brünni ágostonrendi apát (nem szovjet tudós) Tudatos és nagyon alapos kutat") 10.2.2010 genmisk1 1 Áttekintés Mendel és a mendeli törvények Mendel előtt és körül A genetika törvényeinek újbóli felfedezése és a kromoszómák Watson és Crick a molekuláris biológoa központi dogmája 10.2.2010
10.2.2010 genmisk1 1 Áttekintés Mendel és a mendeli törvények Mendel előtt és körül A genetika törvényeinek újbóli felfedezése és a kromoszómák Watson és Crick a molekuláris biológoa központi dogmája 10.2.2010
Spinalis muscularis atrophia (SMA)
") Spinalis muscularis atrophia (SMA) Dr. Sztriha László SZTE ÁOK Gyermekgyógyászati Klinika és Gyermekegészségügyi Központ, Szeged A spinalis muscularis atrophia (SMA) a mucoviscidosis után a leggyakoribb
Spinalis muscularis atrophia (SMA) Dr. Sztriha László SZTE ÁOK Gyermekgyógyászati Klinika és Gyermekegészségügyi Központ, Szeged A spinalis muscularis atrophia (SMA) a mucoviscidosis után a leggyakoribb
Asperger syndrome related suicidal behavior: two case studies Neuropsychiatric Disease and Treatement 2013 (9), 1815-1819
, 1815-1819") Asperger syndrome related suicidal behavior: two case studies Neuropsychiatric Disease and Treatement 2013 (9), 1815-1819 Csupor Éva 2014. október 27. Bevezetés (általában az Asperger-szindrómáról) diagnosztizálás
Asperger syndrome related suicidal behavior: two case studies Neuropsychiatric Disease and Treatement 2013 (9), 1815-1819 Csupor Éva 2014. október 27. Bevezetés (általában az Asperger-szindrómáról) diagnosztizálás
A neurofibromatózis idegrendszeri megnyilvánulása
 A neurofibromatózis idegrendszeri megnyilvánulása Molekuláris Medicina Mindenkinek Fókuszban a Neurofibromatózis Varga Edina Tímea SE Genomikai Medicina és Ritka Betegségek Intézete Neurofibromatózis I.
A neurofibromatózis idegrendszeri megnyilvánulása Molekuláris Medicina Mindenkinek Fókuszban a Neurofibromatózis Varga Edina Tímea SE Genomikai Medicina és Ritka Betegségek Intézete Neurofibromatózis I.
STATISZTIKAI TÜKÖR 2014/126. A népesedési folyamatok társadalmi különbségei. 2014. december 15.
 STATISZTIKAI TÜKÖR A népesedési folyamatok társadalmi különbségei 214/126 214. december 15. Tartalom Bevezető... 1 1. Társadalmi különbségek a gyermekvállalásban... 1 1.1. Iskolai végzettség szerinti különbségek
STATISZTIKAI TÜKÖR A népesedési folyamatok társadalmi különbségei 214/126 214. december 15. Tartalom Bevezető... 1 1. Társadalmi különbségek a gyermekvállalásban... 1 1.1. Iskolai végzettség szerinti különbségek
Dr. Ottó Szabolcs Országos Onkológiai Intézet
 Nemzeti Kutatási és Fejlesztési Program 1. Főirány: Életminőség javítása Nemzeti Onkológiai Kutatás-Fejlesztési Konzorcium a daganatos halálozás csökkentésére 1/48/2001. Részjelentés: 200. November 0.-2004.
Nemzeti Kutatási és Fejlesztési Program 1. Főirány: Életminőség javítása Nemzeti Onkológiai Kutatás-Fejlesztési Konzorcium a daganatos halálozás csökkentésére 1/48/2001. Részjelentés: 200. November 0.-2004.
Genetikai vizsgálat. Intergenomialis kommunikációs zavarok (mtdns depléció/deléció jelenléte esetén javasolt) Alpers syndroma (POLG1 gén analízis)
 Alpers syndroma (POLG1 gén analízis)") Genetikai vizsgálat Intergenomialis kommunikációs zavarok (mtdns depléció/deléció jelenléte esetén javasolt) Alpers syndroma (POLG1 gén analízis) SANDO (POLG1 gén analízis) TWINKLE ANT1 OPA1 SCO2 RRM2B
Genetikai vizsgálat Intergenomialis kommunikációs zavarok (mtdns depléció/deléció jelenléte esetén javasolt) Alpers syndroma (POLG1 gén analízis) SANDO (POLG1 gén analízis) TWINKLE ANT1 OPA1 SCO2 RRM2B
CD8 pozitív primér bőr T-sejtes limfómák 14 eset kapcsán
 CD8 pozitív primér bőr T-sejtes limfómák 14 eset kapcsán Szepesi Ágota 1, Csomor Judit 1, Marschalkó Márta 2 Erős Nóra 2, Kárpáti Sarolta 2, Matolcsy András 1 Semmelweis Egyetem I. Patológiai és Kísérlet
CD8 pozitív primér bőr T-sejtes limfómák 14 eset kapcsán Szepesi Ágota 1, Csomor Judit 1, Marschalkó Márta 2 Erős Nóra 2, Kárpáti Sarolta 2, Matolcsy András 1 Semmelweis Egyetem I. Patológiai és Kísérlet
Kappelmayer János. Malignus hematológiai megbetegedések molekuláris háttere. MOLSZE IX. Nagygyűlése. Bük, 2005 szeptember
 Kappelmayer János Malignus hematológiai megbetegedések molekuláris háttere MOLSZE IX. Nagygyűlése Bük, 2005 szeptember 29-30. Laboratóriumi vizsgálatok hematológiai malignómákban Általános laboratóriumi
Kappelmayer János Malignus hematológiai megbetegedések molekuláris háttere MOLSZE IX. Nagygyűlése Bük, 2005 szeptember 29-30. Laboratóriumi vizsgálatok hematológiai malignómákban Általános laboratóriumi
MUTÁCIÓK. A mutáció az örökítő anyag spontán, maradandó megváltozása, amelynek során új genetikai tulajdonság keletkezik.
 MUTÁCIÓK A mutáció az örökítő anyag spontán, maradandó megváltozása, amelynek során új genetikai tulajdonság keletkezik. Pontmutáció: A kromoszóma egy génjében pár nukleotidnál következik be változás.
MUTÁCIÓK A mutáció az örökítő anyag spontán, maradandó megváltozása, amelynek során új genetikai tulajdonság keletkezik. Pontmutáció: A kromoszóma egy génjében pár nukleotidnál következik be változás.
MI ÁLLHAT A FEJFÁJÁS HÁTTERÉBEN? Dr. HégerJúlia, Dr. BeszterczánPéter, Dr. Deák Veronika, Dr. Szörényi Péter, Dr. Tátrai Ottó, Dr.
 MI ÁLLHAT A FEJFÁJÁS HÁTTERÉBEN? Dr. HégerJúlia, Dr. BeszterczánPéter, Dr. Deák Veronika, Dr. Szörényi Péter, Dr. Tátrai Ottó, Dr. Varga Csaba ESET 46 ÉVES FÉRFI Kórelőzmény kezelt hypertonia kivizsgálás
MI ÁLLHAT A FEJFÁJÁS HÁTTERÉBEN? Dr. HégerJúlia, Dr. BeszterczánPéter, Dr. Deák Veronika, Dr. Szörényi Péter, Dr. Tátrai Ottó, Dr. Varga Csaba ESET 46 ÉVES FÉRFI Kórelőzmény kezelt hypertonia kivizsgálás
Kromoszómák, Gének centromer
 Kromoszómák, Gének A kromoszóma egy hosszú DNS szakasz, amely a sejt életének bizonyos szakaszában (a sejtosztódás előkészítéseként) tömörödik, így fénymikroszkóppal láthatóvá válik. A kromoszómák két
Kromoszómák, Gének A kromoszóma egy hosszú DNS szakasz, amely a sejt életének bizonyos szakaszában (a sejtosztódás előkészítéseként) tömörödik, így fénymikroszkóppal láthatóvá válik. A kromoszómák két
Dr. Máthéné Dr. Szigeti Zsuzsanna és munkatársai
 Kar: TTK Tantárgy: CITOGENETIKA Kód: AOMBCGE3 ECTS Kredit: 3 A tantárgyat oktató intézet: TTK Mikrobiális Biotechnológiai és Sejtbiológiai Tanszék A tantárgy felvételére ajánlott félév: 3. Melyik félévben
Kar: TTK Tantárgy: CITOGENETIKA Kód: AOMBCGE3 ECTS Kredit: 3 A tantárgyat oktató intézet: TTK Mikrobiális Biotechnológiai és Sejtbiológiai Tanszék A tantárgy felvételére ajánlott félév: 3. Melyik félévben
Az anorexia kifejezést először az ókorban használták, eredetileg hiányzó vágyat jelentett,
 Az anorexia kifejezést először az ókorban használták, eredetileg hiányzó vágyat jelentett, később már csak kizárólag az étvágy hiányát jelentette. Kialakulásának magyarázatában kezdetben vallásos elképzelések
Az anorexia kifejezést először az ókorban használták, eredetileg hiányzó vágyat jelentett, később már csak kizárólag az étvágy hiányát jelentette. Kialakulásának magyarázatában kezdetben vallásos elképzelések