exact matching: topics
|
|
|
- Egon Illés
- 6 évvel ezelőtt
- Látták:
Átírás
1 Exact Matching
2 exact matching: topics exact matching search pattern P in text T (P,T strings) Knuth Morris Pratt preprocessing pattern P Aho Corasick pattern of several strings P = { P 1,, P r } Suffix Trees preprocessing text T or several texts database
3 Exact Matching: Preprocessing Pattern(s) Knuth-Morris-Pratt Aho-Corasick
4 KMP example a b a a b a b a a b a a b a b a failure links (suffix = prefix) top: as automaton bottom: as table how to determine? how to use? 6 a b a a b a b a a b a a b a b a 3 7 a b a a b a b a a b a a b a b a 4 8 a b a a b a b a a b a a b... 3
5 KMP computing failure links failure link ~ new best match (after mismatch) òr 0 k-1 a b b a k Flink[1] = 0; for k from 2 to PatLen do fail = Flink[k-1] while ( fail>0 and P[fail] P[k-1] ) do fail = Flink[fail]; od Flink[k] = fail+1; od
6 prefixes via failure links P P 1 P r-1 P k-r+1 P k-1 Flink[k]=r r k P r P 1 P r-1 = P k-r+1 P k-1 maximal r<k all such values r: r 4 r 3 r 2 r 1 k P 1 P r2-1 = P k-r2+1 P k-1 = P r1-r2+1 P r1-1 Flink[r 1 ]=r 2
7 other methods Boyer-Moore T = marktkoopman P = schoenveter schoe work backwards Karp-Rabin fingerprint a i-1 a i a i+n-1 a i+n p 1 p n hash-value a i B n-1 +a i+1 B n-2 +a i+n-1 B 0 a i+1 B n-1 + +a i+n-1 B 1 +a i+n B 0
8 exact matching with a set of patterns P = { P 1,, P r } all occurrences in text T total length m length n AHO CORASICK generalizes KMP failure links longest suffix that is prefix (perhaps in another string) > no subwords within P
9 keyword tree - trie edges ~ letters t a t o t o e p e t r r s y i c e n h c o { potato, poetry, pottery, science, school } o l y e leaves ~ keywords
10 failure links o t a t o p t o a h t t o o e t a t h e r e { potato, tattoo, theater, other } r potato other potato tattoo failure links into other branches! => into other patterns
11 Algorithm for adding the failure links: follow the links existing a a new edge with incoming a follow existing failure links starting at parent node until outgoing a is found
12 Adding failure links o t a t o p a t t o h t o o e t a t h e r e { potato, tattoo, theater, other } r potato other t heater attoo potato ota tattoo breadth first (level-by-level)
13 failure links o t a t o p a t t o h t o o e t a t h e r e { potato, tattoo, theater, other } r shortcuts root to child root (see for example po ) [single letter]
14 preprocessing text T: Suffix Trees
15 trie vs. suffix tree abaab baab aab ab b Text string T (= abaab ) + all its suffixes a b a b a a b a b a a aab b ab aab b b trie suffix tree
16 trie vs. tree a a b b a a b b a a b Trie(T) = O( T ) 2 quadratic bad example: T = a n b n Trie(T) like DFA for the suffixes of T minimize DFA directed acyclic word graph DFA = Deterministic Finite Automaton (also used by Ukkonen) a b ab aab b aab only branching nodes and leaves represented edges labeled by substrings of T correspondence of leaves and suffixes T leaves, hence < T internal nodes Tree(T) = O( T + size(edge labels)) linear
17 nittygritty Order of insertion is position of suffix in T: nittygritty ittygritty ttygritty tygritty ygritty gritty ritty itty tty ty y nittygritty gritty 2 8 ε gritty y t itty ty y gritty gritty ε gritty ε ε
18 nittygritty nittygritty ittygritty ttygritty tygritty ygritty gritty ritty itty tty ty y nittygritty 1-11 gritty ε 2-5 itty gritty ty ε t y 5-5 gritty 6-11 y gritty ε gritty ε implementation: refer to positions of the substrings in T
19 linear time construction nittygritty ittygritty ttygritty tygritty ygritty gritty ritty itty tty ty y Weiner (1973) algorithm of the year McCreight (1976) on-line algorithm (Ukkonen 1992)
20 a Failure link to maximal prefix of suffix in T online construction of suffix trie for T = abaab suffix links b a b abaab baab aab ab εb ε a b a a b a b b a a abaa baa aa εa ε a b b a a a a b b a a b a a next symbol = b from here b already exists
21 application: full text index T pos P pos P in T P is prefix of suffix of T P subtree under P ~ locations of P pos pos
22 example: find itt in nittygritty nittygritty ittygritty ttygritty tygritty ygritty gritty ritty itty tty ty y nittygritty gritty 2 8 ε itty gritty ty ε gritty y 6 t y gritty gritty ε ε 5 11 positions
23 application: longest common substring T P apples plate T pos pos P generalized suffix tree Containing suffixes of both T and T (mark T and T suffixes) pos pos
24 application: counting motifs nittygritty ittygritty ttygritty tygritty ygritty gritty ritty itty tty ty y nittygritty gritty 2 8 ε itty ty 2 gritty 2 ε gritty y 6 t 4 gritty y gritty 2 ε ε 5 11 The suffix trie now contains counts for the number Of suffixes in a sub-trie
25 motif : repeats in DNA as reported by Ukkonen human chromosome 3 the first bases 31 min cpu time (8 processors, 4 GB) human genome: 3x10 9 bases suffix tree for Human Genome feasible
26 longest repeat? Occurrences at: , r Length: 2559 ttagggtacatgtgcacaacgtgcaggtttgttacatatgtatacacgtgccatgatggtgtgctgcacccattaactcgtcatttagcgtta ggtatatctccgaatgctatccctcccccctccccccaccccacaacagtccccggtgtgtgatgttccccttcctgtgtccatgtgttctca ttgttcaattcccacctatgagtgagaacatgcggtgtttggttttttgtccttgcgaaagtttgctgagaatgatggtttccagcttcatccata tccctacaaaggacatgaactcatcatttttttatggctgcatagtattccatggtgtatatgtgccacattttcttaacccagtctacccttgttg gacatctgggttggttccaagtctttgctattgtgaatagtgccgcaataaacatacgtgtgcatgtgtctttatagcagcatgatttataatcc tttgggtatatacccagtaatgggatggctgggtcaaatggtatttctagttctagatccctgaggaatcaccacactgacttccacaatggt tgaactagtttacagtcccagcaacagttcctatttctccacatcctctccagcacctgttgtttcctgactttttaatgatcgccattctaactg gtgtgagatggtatctcattgtggttttgatttgcatttctctgatggccagtgatgatgagcattttttcatgtgttttttggctgcataaatgtctt cttttgagaagtgtctgttcatatccttcgcccacttttgatggggttgtttgtttttttcttgtaaatttgttggagttcattgtagattctgggtatta gccctttgtcagatgagtaggttgcaaaaattttctcccattctgtaggttgcctgttcactctgatggtggtttcttctgctgtgcagaagctct ttagtttaattagatcccatttgtcaattttggcttttgttgccatagcttttggtgttttagacatgaagtccttgcccatgcctatgtcctgaatg gtattgcctaggttttcttctagggtttttatggttttaggtctaacatgtaagtctttaatccatcttgaattaattataaggtgtatattataaggtg taattataaggtgtataattatatattaattataaggtgtatattaattataaggtgtaaggaagggatccagtttcagctttctacatatggctag ccagttttccctgcaccatttattaaatagggaatcctttccccattgcttgtttttgtcaggtttgtcaaagatcagatagttgtagatatgcgg cattatttctgagggctctgttctgttccattggtctatatctctgttttggtaccagtaccatgctgttttggttactgtagccttgtagtatagttt gaagtcaggtagcgtgatggttccagctttgttcttttggcttaggattgacttggcaatgtgggctcttttttggttccatatgaactttaaagt agttttttccaattctgtgaagaaattcattggtagcttgatggggatggcattgaatctataaattaccctgggcagtatggccattttcacaa tattgaatcttcctacccatgagcgtgtactgttcttccatttgtttgtatcctcttttatttcattgagcagtggtttgtagttctccttgaagaggt ccttcacatcccttgtaagttggattcctaggtattttattctctttgaagcaattgtgaatgggagttcactcatgatttgactctctgtttgtctg ttattggtgtataagaatgcttgtgatttttgcacattgattttgtatcctgagactttgctgaagttgcttatcagcttaaggagattttgggctga gacgatggggttttctagatatacaatcatgtcatctgcaaacagggacaatttgacttcctcttttcctaattgaatacccgttatttccctctc ctgcctgattgccctggccagaacttccaacactatgttgaataggagtggtgagagagggcatccctgtcttgtgccagttttcaaaggg aatgcttccagtttttgtccattcagtatgatattggctgtgggtttgtcatagatagctcttattattttgagatacatcccatcaatacctaattt attgagagtttttagcatgaagagttcttgaattttgtcaaaggccttttctgcatcttttgagataatcatgtggtttctgtctttggttctgtttata tgctggagtacgtttattgattttcgtatgttgaaccagccttgcatcccagggatgaagcccacttgatcatggtggataagctttttgatgt gctgctggattcggtttgccagtattttattgaggatttctgcatcgatgttcatcaaggatattggtctaaaattctctttttttgttgtgtctctgt caggctttggtatcaggatgatgctggcctcataaaatgagttagg
27 ten occurrences? ttttttttttttttgagacggagtctcgctctgtcgcccaggctggagtgcagtg gcgggatctcggctcactgcaagctccgcctcccgggttcacgccattct cctgcctcagcctcccaagtagctgggactacaggcgcccgccactacg cccggctaattttttgtatttttagtagagacggggtttcaccgttttagccgg gatggtctcgatctcctgacctcgtgatccgcccgcctcggcctcccaaag tgctgggattacaggcgt Length: 277 Occurrences at: , , , , , In the reversed complement at: , , ,
28 finally suffix tree efficient (linear) storage, but constant 40 large overhead suffix array has constant ±5 hence more practical but has its own complications naïve n log(n) algorithm not too bad
29 suffix array nittygritty ittygritty ttygritty tygritty ygritty gritty ritty itty tty ty y gritty itty ittygritty nittygritty ritty tty ttygritty ty tygritty y ygritty lexicographic order of the suffixes
30 sources Dan Gusfield Algorithms on Strings, Trees, and Sequences Computer Science and Computational Biology lists many applications for suffix trees (and extended implementation details) slides on suffix-trees based on/copied from Esko Ukkonen, Univ Helsinki (Erice School, 30 Oct 2005)
31 Next Generation Sequencing Based on Lecture Notes by R. Shamir [7] E.M. Bakker
32 Introduction Next Generation Technologies The Mapping Problem The MAQ Algorithm The Bowtie Algorithm Burrows-Wheeler Transform Sequence Assembly Problem De Bruijn Graphs Other Assembly Algorithms Overview
33 Introduction 1953 Watson and Crick: the structure of the DNA molecule DNA carrier of the genetic information, the challenge of reading the DNA sequence became central to biological research. methods for DNA sequencing were extremely inefficient, laborious and costly Holley: reliably sequencing the yeast gene for trna Ala required the equivalent of a full year's work per person per base pair (bp) sequenced (1bp/person year) Two classical methods for sequencing DNA fragments by Sanger and Gilbert.
34 Sanger sequencing (sketch): a) polymerase reaction taking place in the ddctp tube. The polymerase extends the labeled primer, randomly incorporating either a normal dctp base or a modified ddctp base. At every position where a ddctp is inserted, polymerization terminates; => a population of fragments; the length of each is a function of the relative distance from the modified base to the primer. b) electrophoretic separation of the products of each of the four reaction tubes above (ddg, dda, ddt, and ddc), run in individual lanes. The bands on the gel represent the respective fragments shown to the right => the complement of the original template (bottom to top)
35 Introduction 1980 In the 1980s methods were augmented by partial automation the cloning method, which allowed fast and exponential replication of a DNA fragment Start of the human genome project: sequencing efficiency reached 200,000 bp/person-year End of the human genome project: 50,000,000 bp/person-year. (Total cost summed to $3 billion.) Note: Moore s Law doubling transistor count every 2 years Here: doubling of #base pairs/person-year every 1.5 years
36 Introduction Recently ( /2012): new sequencing technologies next generation sequencing (NGS) or deep sequencing reliable sequencing of 100x10 9 bp/person-year. NGS now allows compiling the full DNA sequence of a person for ~ $10,000 within 3-5 years cost is expected to drop to ~$1000. Currently (2013) full humane genome scan with a coverage of 3 for around 2000 euro.
37 Next Generation Sequencing Technologies Next Generation Sequencing technology of Illumina, one of the leading companies in the field: DNA is replicated => millions of copies. All DNA copies are randomly shredded into various fragments using restriction enzymes or mechanical means. The fragments form the input for the sequencing phase. Hundreds of copies of each fragment are generated in one spot (cluster) on the surface of a huge matrix. Initially it is not known which fragment sits where. Note: There are quite a few other available technologies available and currently under development.
38 Next Generation Sequencing technology of Illumina (continued): A DNA replication enzyme is added to the matrix along with 4 slightly modified nucleotides. The 4 nucleotides are slightly modified chemically so that: each would emit a unique color when excited by laser, each one would terminate the replication. Hence, the growing complementary DNA strand on each fragment in each cluster is extended by one nucleotide at a time.
39 Next Generation Sequencing technology of Illumina (continued): A laser is used to obtain a read of the actual nucleotide that is added in each cluster. (The multiple copies in a cluster provide an amplified signal.) The solution is washed away together with the chemical modification on the last nucleotide which prevented further elongation and emitted the unique signal. Millions of fragments are sequenced efficiently and in parallel. The resulting fragment sequences are called reads. Reads form the input for further sequence mapping and assembly.
 A laser is used to obtain a read of the nucleotide just added. (3) The full sequence of a fragment thus determined through successive iterations of the process.")
40 Next Generation Sequencing Technologies (1) A modified nucleotide is added to the complementary DNA strand by a DNA polymerase enzyme. (2) A laser is used to obtain a read of the nucleotide just added. (3) The full sequence of a fragment thus determined through successive iterations of the process. (4) A visualization of the matrix where fragment clusters are attached to flow cells.
41 The Mapping Problem The Short Read Mapping Problem INPUT: m l-long reads S 1,, S m and an approximate reference genome R. QUESTION: What are the positions x 1,, x m along R where each read matches? For example: after sequencing the genome of a person we want to map it to an existing sequence of the human genome. The new sample will not be 100% identical to the reference genome: natural variation in the population some reads may align to their position in the reference with mismatches or gaps sequencing errors repetitive regions in the genome make it difficult to decide where to map the read Humans are diploid organisms
42 Possible Solutions for the Mapping Problem The problem of aligning a short read to a long genome sequence is exactly the problem of local alignment. But in case of the human genome: the number of reads m is usually the length of a read l is bp the length of the genome R is 3x 10 9 bp (or twice, for the diploid genome).
43 Solutions for the Mapping Problem Naive algorithm scan the reference string R for each S i, matching the read at each position p and picking the best match. Time complexity: O(ml R ) for exact or inexact matching. m number of reads, l read length Considering the parameters for our problem, this is clearly impractical. Less naive solution use the Knuth-Morris-Pratt algorithm to match each S i to R Time complexity: O(m(l+ R )) = O(ml + m R ) for exact matching. A substantial improvement but still not enough
44 The Mapping Problem: Suffix Trees 1. Build a suffix tree for R. 2. For each S i (i in {1,,m})find matches by traversing the tree from the root. Time complexity: O(ml+ R ), assuming the tree is built using Ukkonen's linear-time algorithm. Notes: Time complexity is practical. we only need to build the tree for the reference genome once. The tree can be saved and used repeatedly for mapping new sequences, until an updated version of the genome is issued.
45 The Mapping Problem: Suffix Trees Space complexity now becomes the obstacle: The leaves of the suffix tree also hold the indices where the suffixes begin => the tree requires O( R log R ) bits just for the binary coding of the indices. The constants are large due to the additional layers of information required for the tree (such as suffix links, etc.). The original text demands just R log(#symbols) bits => we can store the human genome using ~750MB, but we need ~64GB for the tree (2013: doable)! Final problem: suffix trees allow only for exact matching.
46 The Mapping Problem: Hashing Preprocess the reference genome R into a hash table H. where the keys of the hash are all the substrings of length l in R, and Table H contains: the position p in R where the substring ends. Matching: given S i the algorithm returns H(S i ). Time complexity: O(ml+l R ) Note: The space complexity is O(l R + R log R ) since we must also hold the binary representation of each substring's position. (Still too high.) Packing the substrings into bit-vectors representing each nucleotide as a 2-bit code reduces the space complexity by a factor of 4. Practical solution: Partition the genome into several chunks, each of the size of the cache memory, and run the algorithm on each chunk in turn. Again, only exact matching allowed.
47 The Mapping Problem: The MAQ Algorithm The MAQ (Mapping and Alignment with Qualities) algorithm by Li, Ruan and Durbin, 2008 [5] An efficient solution to the short read mapping problem Allows for inexact matching, up to a predetermined maximum number of mismatches. Memory efficient. Measures for base and read quality, and for identifying sequence variants.
48 The MAQ Algorithm A key insight: If a read in its correct position to the reference genome has one mismatch, then partitioning that read into two makes sure one part is still exactly matched. Hence, if we perform an exact match twice, each time checking only a subset of the read bases (called a template), one subset is enough to find the match. For 2 mismatches, it is possible to create six templates: some are noncontiguous each template covers half the read Each template has a partner template that complements it to form the full read If the read has no more than two mismatches, at least one template will not be affected by any mismatch. Twenty templates guarantee a fully matched template for any 3- mismatched read, etc.

49 The MAQ Algorithm: Templates A and B are reads; purple boxes indicate positions of mismatches with respect to the reference. The numbered templates have blue boxes in the positions they cover. 1 mismatch: comparing read A through template 1 => no full match, but template 2 fully matched. 2 mismatches: read B is fully matched with template 6 despite this. Any combination of up to two mismatched positions will be avoided by at least one of the six templates. Note: six templates guarantee 57% of all 3 mismatches will also have at least one unaffected template.
50 The MAQ Algorithm The algorithm: generate the number of templates required to guarantee at least one full match for the desired maximal number of mismatches, and use that exact match as a seed for extending across the full read. Identifying the exact matches is done by hashing the read templates into template-specific hash tables, and scanning the reference genome R against each table. Note: It is not necessary to generate templates and hash tables for the full read, since the initial seed will undergo extension, e.g. using the Smith- Waterman algorithm. Therefore the algorithm initially processes only the first 28 bases of each read. (The first bases are the most accurate bases of the reads.)
51 The MAQ Algorithm Algorithm (for the case of up to two mismatches): 1. Index the first 28 bases of each read with complementary templates 1, 2; generate hash tables H1, H2. 2. Scan the reference: for each position and each orientation, query a 28-bp window through templates 1, 2 against the appropriate tables H1, H2, respectively. If hit, extend and score the complete read based on mismatches. For each read, keep the two best scoring hits and the number of mismatches therein. 3. Repeat steps 1+2 with complementary templates 3, 4, then 5, 6. Remark: The reason for indexing against a pair of complementary templates each time has to do with a feature of the algorithm regarding paired-end reads (see original paper).
52 The MAQ Algorithm Complexity Time Complexity: O(ml) for generating the hash tables in step 1, and O(l R ) for scanning the genome in step 2. We repeat steps 1+2 three times in this implementation but that does not affect the asymptotic complexity. Space Complexity: O(ml) for holding the hash tables in step 1, and O(ml+ R ) total space for step 2, but only O(ml) space in main memory at any one time.
53 The MAQ Algorithm Complexity Not the full read length l is used, but rather a window of length l' <= l (28-bp in the implementation presented above). The size of each key in a template hash table is only l' /2, since each template covers half the window. => More precisely time and space complexity of step 1 is: O(2 m l' /2) = O(ml' ). This reduction in space makes running the algorithm with the cache memory of a desktop computer feasible. The time and space required for extending a match in step 2 using the Smith- Waterman algorithm, where p the probability of a hit (using the l length window): The time complexity : O(l' R + p R l 2 ). The space complexity: O(ml' + l 2 ). Note: The value of p is small, and decreases drastically the longer l' is, since most l' -long windows in the reference genome will likely not capture the exact coordinates of a true read.
54 Read Mapping Qualities The MAQ algorithm provides a measure of the confidence level for the mapping of each read, denoted by QS and defined as: QS = -10 log 10 (Pr{read S is wrongly mapped}) For example: QS = 30 if the probability of incorrect mapping of read S is 1/1000. This confidence measure is called phred-scaled quality, following the scaling scheme originally introduced by Phil Green and colleagues[3] for the human genome project.
55 QS Confidence Levels A simplified statistical model for inferring QS: Assume we know the reads are supposed to match the reference precisely. So any mismatches must be the result of sequencing errors. Assuming that sequencing errors along a read are independent, we can define: the probability p(z x, u) of obtaining the read z given that its position in reference x is u as the product of the error probabilities of the observed mismatches in the alignment of the read at that position. These base error probabilities are empirically determined during the sequencing phase based on the characteristics of the emitted signal at each step.
56 QS Example If read z aligned at position u yields 2 mismatches that have a phred-scaled base quality of 10 and 20, then: p(z x, u) = 10 -(20+10)/10 = Note that we will be able to calculate the posterior probability p s (u x, z) of position u being the correct mapping for an observed read z using Bayes' theorem (see text). Being able to calculate these posterior probability, we can now give our confidence measure as: Q s (u x, z) = -10 log 10 (1 - p s (u x, z)) Note: MAQ uses a more sophisticated statistical model to deal with the issues we neglected in this simplification, such as true mismatches that are not the result of sequencing errors (SNPs = single nucleotide polymorphisms).
57 The Bowtie Algorithm We have seen that one way to map reads to a reference genome is to index into a hash table either all l-long windows of the genome or of the reads requiring a lot of space. The Bowtie algorithm, presented in 2009 by Langmead et al.[1], solves the problem through a more space-efficient indexing scheme. The indexing scheme used is called the Burrows-Wheeler transform[2] and was originally developed for data compression purposes.
58 Burrows-Wheeler Transform Applying the Burrows-Wheeler transform BW(T) to the text T = "the next text that i index.": 1. First, we generate all cyclic shifts of T. 2. Next, we sort these shifts lexicographically. define the character '.' as the minimum and we assume that it appears exactly once, as the last symbol in the text. followed lexicographically by ' followed by the English letters according to their natural ordering. Call the resulting matrix M. The transform BW(T) is defined as the sequence of the last characters in the rows of M. Note that the last column is a permutation of all characters in the text since each character appears in the last position in exactly one cyclic shift.
59 Burrows-Wheeler Transform Some of the cyclic shifts of T sorted lexicographically and indexed by the last character.
60 Burrows-Wheeler Transform Storing BW(T) requires the same space as the size of the text T since it is simply a permutation of T. In the case of the human genome, each character can be represented by 2 bits => storing the permutation requires 2 times 3x10 9 bits (instead of ~30 times 3x10 9 for storing all indices of T).
61 Burrows-Wheeler Transform Note: we can infer the following information from BW(T): 1. The number of occurrences in T of the character 'e' = counting the number of occurrences of 'e in BW(T) (permutation of the text). 2. The first column of the matrix M can be obtained by sorting BW(T) lexicographically. 3. The number of occurrences of the substring 'xt' in T: BW(T) is the last column of the lexicographical sorting of the shifts. The character at the last position of a row appears in the text T immediately prior to the first character in the same row (each row is a cyclical shift). => consider the interval of 't' in the first column, and check whether any of these rows have an 'x at the last position.
 by sorting the last column.")
62 Sorted BW(T) BW(T) Recovering the first column (left) by sorting the last column.
63 Burrows-Wheeler Transform After transforming a text T into BW(T) also the second column can be derived: as we saw that 'xt' appears twice in the text, and we see that 3 rows start with an 'x'. Two of those must be followed by a 't, where the lexicographical sorting determines which ones. Another 'x' is followed by a '.' (see first row) => '.' must follow the first 'x' in the first column since '.' is smaller lexicographically than 't'. The second and third occurrences of 'x' in the first column are therefore followed by 't'. Note: We can use the same process to recover the characters at the second column for each interval, and then the third, etc. t text that i index. the nex t that i index. the next tex text that i index.the next that i index. the next text the next text that i index.
64 F L Last-first mapping: Each 't' character in L is linked to its position in F and no crossed links are possible. The j-th occurrence of character X in L corresponds to the same text character as the j-th occurrence of X in F.
65 Burrows-Wheeler Transform The previous two central properties of the BW-transform are captured in the Lemma by Ferragina and Manzini[4]: Lemma 12.1 (Last-First Mapping): Let M be the matrix whose rows are all cyclical shifts of T sorted lexicographically, and let L(i) be the character at the last column of row i and F(i) be the first character in that row. Then: 1. In row i of M, L(i) precedes F(i) in the original text: T = L(i) F(i) 2. The j-th occurrence of character X in L corresponds to the same text character as the j-th occurrence of X in F.
66 F L Last-first mapping: j-th occurrence of character 't' character in L is linked to its j-th position in F and no crossed links are possible.
67 Burrows-Wheeler Transform Proof: 1. Follows directly from the fact that each row in M is a cyclical shift. 2. Let X j denote the j-th occurrence of char X in L, and let α be the character following X j in the text and β the character following X j +1. Then, since X j appears above X j +1 in L, α must be equal or lexicographically smaller than β. This is true since the order is determined by lexicographical sorting of the full row and the character in F follows the one in L. Hence, when character X j appears in F, it will again be above X j +1, since α and β now appear in the second column and X α lexicographically X β.
68 Reconstructing the Text Algorithm UNPERMUTE for reconstructing a text T from its Burrows-Wheeler transform BW(T) utilizing Lemma 12.1 [4]: assume the actual text T is of length u append a unique $ character at the end, which is the smallest lexicographically UNPERMUTE[BW(T)] 1. Compute the array C[1,, Σ ] : where C(c) is the number of characters {$, 1,,c-1} in T, i.e., the number of characters that are lexicographically smaller than c 2. Construct the last-first mapping LF, tracing every character in L to its corresponding position in F: LF[i] = C(L[i]) + r(l[i], i) + 1, where r(c, i) is the number of occurrences of character c in the prefix L[1, i -1] 3. Reconstruct T backwards: s = 1, T(u) = L[1]; for i = u 1,, 1 do s = LF[s]; T[i] = L[s]; od;

69 Notice, that C(e) + 1 = 9 is the position of the first occurrence of e' in F. C F L Last-first mapping: Each 't' character in L is linked to its position in F and no crossed links are possible.
70 Example T = acaacg$ (u = 6) is transformed to BW(T) = gc$aaac, and we now wish to reconstruct T from BW(T) using UNPERMUTE: 1. Compute array C. For example: C(c) = 4 since there are 4 occurrences of characters smaller than 'c' in T ('$' and 3 occurrences of 'a'). Now C(c) + 1 = 5 is the position of the first occurrence of 'c' in F. 2. Perform the LF mapping. For example, LF[c2] = C(c) + r(c,7) + 1 = 6, and indeed the second occurrence of 'c' in F is at F[6]. 3. Determine the last character in T: T(6) = L(1) = 'g'. 4. Iterate backwards over all positions using the LF mapping. For example, to recover the character T(5), we use the LF mapping to trace L(1) to F(7), and then T(5) = L(7) = 'c'. Remark We do not actually hold F in memory, we only keep the array C defined above, of size Σ, which we can easily obtained from L.

71 Determine its location in F and find its corresponding predecessor in L using C(.) and r(.,.) i.e., find corresponding g using the last-first mapping First Last Example of running UNPERMUTE to recover the original text. Source: [1].
72 Exact Matching EXACTMATCH exact matching of a query string P to T, given BW(T)[4] is similar to UNPERMUTE, we use the same C and r(c, i) denote by sp the position of the first row in the interval of rows in M we are currently considering denote by ep the position of the first row beyond this interval of rows => the interval of rows is defined by the rows sp,, ep - 1. EXACTMATCH[P[1,,p], BW(T)] 1. c = P[p]; sp = C[c] + 1; ep = C[c+1] + 1; i = p - 1; 2. while sp < ep and i >= 1 c = P[i]; sp = C[c] + r(c, sp) + 1; ep = C[c] + r(c, ep) + 1; i = i - 1; 3. if (sp == ep) return "no match"; else return sp, ep;

73 P = aac Example of running EXACTMATCH to find a query string in the text. Source: [1].
74 Example Use T = acaacg while searching for P = 'aac': 1. First, we initialize sp and ep to define the interval of rows beginning with the last character in P, which is 'c': sp = C(c) + 1 = 5. ep = C(g) + 1 = Next, we consider the preceding character in P, namely 'a'. We redefine the interval as the rows that begin with 'ac' utilizing the LF mapping. Specifically: sp = C(a) + r(a, 5) + 1 = = 3. ep = C(a) + r(a, 7) + 1 = = Now, we consider the next preceding character, namely 'a'. We now redefine the interval as the rows that begin with 'aac'. Specifically: sp = C(a) + r(a, 3) + 1 = = 2. ep = C(a) + r(a, 5) + 1 = = Having thus covered all positions of P, we return the final interval calculated (whose size equals the number of occurrences of P in T).
75 Exact Matching EXACTMATCH returns the indices of rows in M that begin with the query it does not provide the offset of each match in T. If we kept the position in T corresponding to the start of each row we would waste a lot of space. we can mark only some rows with pre-calculated offsets. Then, if the row where EXACTMATCH found the query has this offset, we can return it immediately. Or use LF mapping to find a row that has a precalculated offset, and add the number of times the procedure is applied to obtain the position in which we are interested. => time-space trade-of associated with this process.
76 Inexact Matching Sketch Each character in a read has a numeric quality value, with lower values indicating a higher likelihood of a sequencing error. Similar to EXACTMATCH, calculating matrix intervals for successively longer query suffixes. If the range becomes empty (a suffix does not occur in the text), then the algorithm selects an already-matched query position and substitute a different base there, introducing a mismatch into the alignment. The EXACTMATCH search resumes from just after the substituted position. The algorithm selects only those substitutions that are consistent with the alignment policy and yield a modified suffix that occurs at least once in the text. If there are multiple candidate substitution positions, then the algorithm greedily selects a position with a maximal quality value.
77 The Assembly Problem How to assemble an unknown genome based on many highly overlapping short reads from it? Problem Sequence assembly INPUT: m l-long reads S 1 ; : : : ; S m. QUESTION: What is the sequence of the full genome? The crucial difference between the problems of mapping and assembly is that now we do not have a reference genome, and we must assemble the full sequence directly from the reads.
78 De Bruijn Graphs Definition A k-dimensional de Bruijn graph of n symbols is a directed graph representing overlaps between sequences of symbols. It has n k vertices, consisting of all possible k-tuples of the given symbols. The same symbol may appear multiple times in a tuple. If we have the set of symbols A = {a 1,,a n } then the set of vertices is: V = { (a 1,, a 1,a 1 ), (a 1,, a 1 ; a 2 ),,(a 1,, a 1, a n ); (a 1,, a 2, a 1 ),, (a n,, a n, a n )} If a vertice w can be expressed by shifting all symbols of another vertex v by one place to the left and adding a new symbol at the end, then v has a directed edge to w. Thus the set of directed edges is: E = {( (v 1, v 2,, v k ), (w 1,w 2,, w k ) ) v 2 = w 1, v 3 = w 2,, v k = w k-1, and w k new}
79 A portion of a 4-dimensional de Bruijn graph, the vertex CAAA has a directed edge to vertex AAAC, because if we shift the label CAAA to the left and add the symbol C we get the label AAAC.
80 De Bruijn Graphs Given a read, every contiguous (k+1)-long word in it corresponds to an edge in the de Bruijn graph. Form a subgraph G of the full de Bruijn graph by introducing only the edges that correspond to (k+1)-long words in some read. A path in this graph defines a potential subsequence in the genome. Hence, we can convert a read to its corresponding path in the constructed subgraph G

81 Using the graph from the previous slide, we construct the path corresponding to CCAACAAAAC: shift the sequence one position to the left each time, and marking the vertex with the label of the 4 first nucleotides.
82 De Bruijn Graphs Form paths for all the reads Identify common vertices on different paths Merge different read-paths through these common vertices of the paths into one long sequence
83 The two reads in (a) are converted to paths in the graph The common vertex TGAG is identified Combine these two paths into (b).
84 De Bruijn Graphs Velvet, presented in 2008 by Zerbino and Birney[6], is an algorithm which utilizes de Bruijn graphs to assemble reads in this way. However, this assembly process can encounter some difficulties: repeats in the genome will manifest themselves as cycles in the merged path that we form. It is impossible to tell how many times we must traverse each cycle in order to form the full sequence of the genome. Even worse, if we have two cycles starting at the same vertex, we cannot tell which one to traverse first. Velvet attempts to address this issue by utilizing the extra information we have in the case of paired-ends sequencing.
85 Other Assembly Algorithms HMM based Majority based Etc.
86 Bibliography [1] Ben Langmead, Cole Trapnell, Mihai Pop and Steven L. Salzberg. Ultrafast and memory-effcient alignment of short DNA sequences to the human genome. Genome Biology, [2] Michael Burrows and David Wheeler. A block sorting lossless data compression algorithm. Technical Report 124, Digital Equipment Corporation, [3] Brent Ewing and Phil Green. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Research, [4] Paulo Ferragina and Giovani Manzini. Opportunistic data structures with applications. FOCS '00 Proceedings of the 41st Annual Symposium on Foundations of Computer Science, [5] Heng Li, Jue Ruan and Richard Durbin. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Research, [6] Daniel R. Zerbino and Ewan Birney. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Research, [7] Ron Shamir, Computational Genomics Fall Semester, 2010 Lecture 12: Algorithms for Next Generation Sequencing Data, January 6, 2011 Scribe: Anat Gluzman and Eran Mick
87 Appendix Some basic terminology and techniques
88 DNA Replication DNA Polymerase is a family of enzymes carrying out DNA replication. A Primer is a short fragment of DNA or RNA that pairs with a template DNA. Polymerase Chain Reaction Mixture of templates and primers is heated such that templates separate in strands Uses a pair of primers spanning a target region in template DNA Polymerizes partner strands in each direction from the primers using thermo stable DNA polymerase repeat
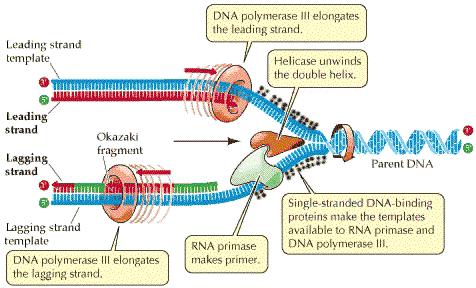
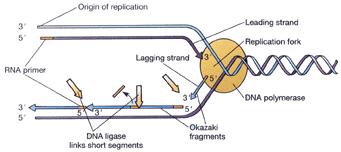
89 DNA Replication Enzyme
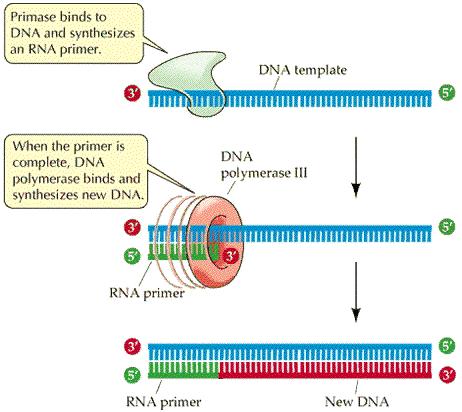
90 Primers
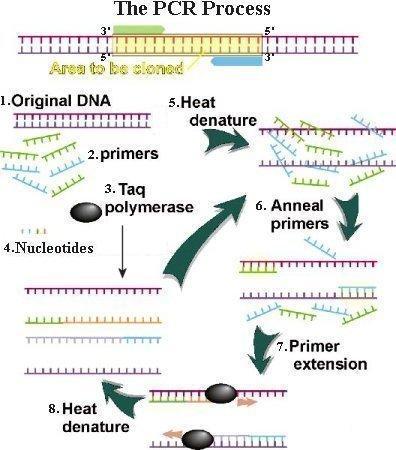
91
92 DNA Restriction DNA restriction enzymes Recognize a specific nucleotide sequence (4 8 bp long, often palindromic) Make a double-stranded DNA cut For example HaeIII recognizes the following sequences and cuts between adjacent G and C giving a blunt end: 5 GGCC3 3 CCGG5 Also restriction endonuclease (restriction enzymes) that cut with an offset and thus producing sticky ends are possible.
93 Restriction Enzymes
Mapping Sequencing Reads to a Reference Genome
 Mapping Sequencing Reads to a Reference Genome High Throughput Sequencing RN Example applications: Sequencing a genome (DN) Sequencing a transcriptome and gene expression studies (RN) ChIP (chromatin immunoprecipitation)
Mapping Sequencing Reads to a Reference Genome High Throughput Sequencing RN Example applications: Sequencing a genome (DN) Sequencing a transcriptome and gene expression studies (RN) ChIP (chromatin immunoprecipitation)
Construction of a cube given with its centre and a sideline
 Transformation of a plane of projection Construction of a cube given with its centre and a sideline Exercise. Given the center O and a sideline e of a cube, where e is a vertical line. Construct the projections
Transformation of a plane of projection Construction of a cube given with its centre and a sideline Exercise. Given the center O and a sideline e of a cube, where e is a vertical line. Construct the projections
Genome 373: Hidden Markov Models I. Doug Fowler
 Genome 373: Hidden Markov Models I Doug Fowler Review From Gene Prediction I transcriptional start site G open reading frame transcriptional termination site promoter 5 untranslated region 3 untranslated
Genome 373: Hidden Markov Models I Doug Fowler Review From Gene Prediction I transcriptional start site G open reading frame transcriptional termination site promoter 5 untranslated region 3 untranslated
On The Number Of Slim Semimodular Lattices
 On The Number Of Slim Semimodular Lattices Gábor Czédli, Tamás Dékány, László Ozsvárt, Nóra Szakács, Balázs Udvari Bolyai Institute, University of Szeged Conference on Universal Algebra and Lattice Theory
On The Number Of Slim Semimodular Lattices Gábor Czédli, Tamás Dékány, László Ozsvárt, Nóra Szakács, Balázs Udvari Bolyai Institute, University of Szeged Conference on Universal Algebra and Lattice Theory
Cluster Analysis. Potyó László
 Cluster Analysis Potyó László What is Cluster Analysis? Cluster: a collection of data objects Similar to one another within the same cluster Dissimilar to the objects in other clusters Cluster analysis
Cluster Analysis Potyó László What is Cluster Analysis? Cluster: a collection of data objects Similar to one another within the same cluster Dissimilar to the objects in other clusters Cluster analysis
Lecture 11: Genetic Algorithms
 Lecture 11 1 Linear and Combinatorial Optimization Lecture 11: Genetic Algorithms Genetic Algorithms - idea Genetic Algorithms - implementation and examples Lecture 11 2 Genetic algorithms Algorithm is
Lecture 11 1 Linear and Combinatorial Optimization Lecture 11: Genetic Algorithms Genetic Algorithms - idea Genetic Algorithms - implementation and examples Lecture 11 2 Genetic algorithms Algorithm is
Miskolci Egyetem Gazdaságtudományi Kar Üzleti Információgazdálkodási és Módszertani Intézet. Hypothesis Testing. Petra Petrovics.
 Hypothesis Testing Petra Petrovics PhD Student Inference from the Sample to the Population Estimation Hypothesis Testing Estimation: how can we determine the value of an unknown parameter of a population
Hypothesis Testing Petra Petrovics PhD Student Inference from the Sample to the Population Estimation Hypothesis Testing Estimation: how can we determine the value of an unknown parameter of a population
Statistical Inference
 Petra Petrovics Statistical Inference 1 st lecture Descriptive Statistics Inferential - it is concerned only with collecting and describing data Population - it is used when tentative conclusions about
Petra Petrovics Statistical Inference 1 st lecture Descriptive Statistics Inferential - it is concerned only with collecting and describing data Population - it is used when tentative conclusions about
Correlation & Linear Regression in SPSS
 Petra Petrovics Correlation & Linear Regression in SPSS 4 th seminar Types of dependence association between two nominal data mixed between a nominal and a ratio data correlation among ratio data Correlation
Petra Petrovics Correlation & Linear Regression in SPSS 4 th seminar Types of dependence association between two nominal data mixed between a nominal and a ratio data correlation among ratio data Correlation
Miskolci Egyetem Gazdaságtudományi Kar Üzleti Információgazdálkodási és Módszertani Intézet Nonparametric Tests
 Nonparametric Tests Petra Petrovics Hypothesis Testing Parametric Tests Mean of a population Population proportion Population Standard Deviation Nonparametric Tests Test for Independence Analysis of Variance
Nonparametric Tests Petra Petrovics Hypothesis Testing Parametric Tests Mean of a population Population proportion Population Standard Deviation Nonparametric Tests Test for Independence Analysis of Variance
Performance Modeling of Intelligent Car Parking Systems
 Performance Modeling of Intelligent Car Parking Systems Károly Farkas Gábor Horváth András Mészáros Miklós Telek Technical University of Budapest, Hungary EPEW 2014, Florence, Italy Outline Intelligent
Performance Modeling of Intelligent Car Parking Systems Károly Farkas Gábor Horváth András Mészáros Miklós Telek Technical University of Budapest, Hungary EPEW 2014, Florence, Italy Outline Intelligent
Using the CW-Net in a user defined IP network
 Using the CW-Net in a user defined IP network Data transmission and device control through IP platform CW-Net Basically, CableWorld's CW-Net operates in the 10.123.13.xxx IP address range. User Defined
Using the CW-Net in a user defined IP network Data transmission and device control through IP platform CW-Net Basically, CableWorld's CW-Net operates in the 10.123.13.xxx IP address range. User Defined
Dependency preservation
 Adatbázis-kezelés. (4 előadás: Relácó felbontásai (dekomponálás)) 1 Getting lossless decomposition is necessary. But of course, we also want to keep dependencies, since losing a dependency means, that
Adatbázis-kezelés. (4 előadás: Relácó felbontásai (dekomponálás)) 1 Getting lossless decomposition is necessary. But of course, we also want to keep dependencies, since losing a dependency means, that
Computer Architecture
 Computer Architecture Locality-aware programming 2016. április 27. Budapest Gábor Horváth associate professor BUTE Department of Telecommunications ghorvath@hit.bme.hu Számítógép Architektúrák Horváth
Computer Architecture Locality-aware programming 2016. április 27. Budapest Gábor Horváth associate professor BUTE Department of Telecommunications ghorvath@hit.bme.hu Számítógép Architektúrák Horváth
Supplementary Figure 1
 Supplementary Figure 1 Plot of delta-afe of sequence variants detected between resistant and susceptible pool over the genome sequence of the WB42 line of B. vulgaris ssp. maritima. The delta-afe values
Supplementary Figure 1 Plot of delta-afe of sequence variants detected between resistant and susceptible pool over the genome sequence of the WB42 line of B. vulgaris ssp. maritima. The delta-afe values
discosnp demo - Peterlongo Pierre 1 DISCOSNP++: Live demo
 discosnp demo - Peterlongo Pierre 1 DISCOSNP++: Live demo Download and install discosnp demo - Peterlongo Pierre 3 Download web page: github.com/gatb/discosnp Chose latest release (2.2.10 today) discosnp
discosnp demo - Peterlongo Pierre 1 DISCOSNP++: Live demo Download and install discosnp demo - Peterlongo Pierre 3 Download web page: github.com/gatb/discosnp Chose latest release (2.2.10 today) discosnp
Supporting Information
 Supporting Information Cell-free GFP simulations Cell-free simulations of degfp production were consistent with experimental measurements (Fig. S1). Dual emmission GFP was produced under a P70a promoter
Supporting Information Cell-free GFP simulations Cell-free simulations of degfp production were consistent with experimental measurements (Fig. S1). Dual emmission GFP was produced under a P70a promoter
Széchenyi István Egyetem www.sze.hu/~herno
 Oldal: 1/6 A feladat során megismerkedünk a C# és a LabVIEW összekapcsolásának egy lehetőségével, pontosabban nagyon egyszerű C#- ban írt kódból fordítunk DLL-t, amit meghívunk LabVIEW-ból. Az eljárás
Oldal: 1/6 A feladat során megismerkedünk a C# és a LabVIEW összekapcsolásának egy lehetőségével, pontosabban nagyon egyszerű C#- ban írt kódból fordítunk DLL-t, amit meghívunk LabVIEW-ból. Az eljárás
SOLiD Technology. library preparation & Sequencing Chemistry (sequencing by ligation!) Imaging and analysis. Application specific sample preparation
 Imaging and analysis. Application specific sample preparation") SOLiD Technology Application specific sample preparation Application specific data analysis library preparation & emulsion PCR Sequencing Chemistry (sequencing by ligation!) Imaging and analysis SOLiD
SOLiD Technology Application specific sample preparation Application specific data analysis library preparation & emulsion PCR Sequencing Chemistry (sequencing by ligation!) Imaging and analysis SOLiD
Véges szavak általánosított részszó-bonyolultsága
 Véges szavak általánosított részszó-bonyolultsága KÁSA Zoltán Sapientia Erdélyi Magyar Tudományegyetem Kolozsvár Marosvásárhely Csíkszereda Matematika-Informatika Tanszék, Marosvásárhely Budapest, 2010.
Véges szavak általánosított részszó-bonyolultsága KÁSA Zoltán Sapientia Erdélyi Magyar Tudományegyetem Kolozsvár Marosvásárhely Csíkszereda Matematika-Informatika Tanszék, Marosvásárhely Budapest, 2010.
Ensemble Kalman Filters Part 1: The basics
 Ensemble Kalman Filters Part 1: The basics Peter Jan van Leeuwen Data Assimilation Research Centre DARC University of Reading p.j.vanleeuwen@reading.ac.uk Model: 10 9 unknowns P[u(x1),u(x2),T(x3),.. Observations:
Ensemble Kalman Filters Part 1: The basics Peter Jan van Leeuwen Data Assimilation Research Centre DARC University of Reading p.j.vanleeuwen@reading.ac.uk Model: 10 9 unknowns P[u(x1),u(x2),T(x3),.. Observations:
Correlation & Linear Regression in SPSS
 Correlation & Linear Regression in SPSS Types of dependence association between two nominal data mixed between a nominal and a ratio data correlation among ratio data Exercise 1 - Correlation File / Open
Correlation & Linear Regression in SPSS Types of dependence association between two nominal data mixed between a nominal and a ratio data correlation among ratio data Exercise 1 - Correlation File / Open
FAMILY STRUCTURES THROUGH THE LIFE CYCLE
 FAMILY STRUCTURES THROUGH THE LIFE CYCLE István Harcsa Judit Monostori A magyar társadalom 2012-ben: trendek és perspektívák EU összehasonlításban Budapest, 2012 november 22-23 Introduction Factors which
FAMILY STRUCTURES THROUGH THE LIFE CYCLE István Harcsa Judit Monostori A magyar társadalom 2012-ben: trendek és perspektívák EU összehasonlításban Budapest, 2012 november 22-23 Introduction Factors which
Angol Középfokú Nyelvvizsgázók Bibliája: Nyelvtani összefoglalás, 30 kidolgozott szóbeli tétel, esszé és minta levelek + rendhagyó igék jelentéssel
 Angol Középfokú Nyelvvizsgázók Bibliája: Nyelvtani összefoglalás, 30 kidolgozott szóbeli tétel, esszé és minta levelek + rendhagyó igék jelentéssel Timea Farkas Click here if your download doesn"t start
Angol Középfokú Nyelvvizsgázók Bibliája: Nyelvtani összefoglalás, 30 kidolgozott szóbeli tétel, esszé és minta levelek + rendhagyó igék jelentéssel Timea Farkas Click here if your download doesn"t start
Adatbázisok 1. Rekurzió a Datalogban és SQL-99
 Adatbázisok 1 Rekurzió a Datalogban és SQL-99 Expressive Power of Datalog Without recursion, Datalog can express all and only the queries of core relational algebra. The same as SQL select-from-where,
Adatbázisok 1 Rekurzió a Datalogban és SQL-99 Expressive Power of Datalog Without recursion, Datalog can express all and only the queries of core relational algebra. The same as SQL select-from-where,
Miskolci Egyetem Gazdaságtudományi Kar Üzleti Információgazdálkodási és Módszertani Intézet. Nonparametric Tests. Petra Petrovics.
 Nonparametric Tests Petra Petrovics PhD Student Hypothesis Testing Parametric Tests Mean o a population Population proportion Population Standard Deviation Nonparametric Tests Test or Independence Analysis
Nonparametric Tests Petra Petrovics PhD Student Hypothesis Testing Parametric Tests Mean o a population Population proportion Population Standard Deviation Nonparametric Tests Test or Independence Analysis
Phenotype. Genotype. It is like any other experiment! What is a bioinformatics experiment? Remember the Goal. Infectious Disease Paradigm
 It is like any other experiment! What is a bioinformatics experiment? You need to know your data/input sources You need to understand your methods and their assumptions You need a plan to get from point
It is like any other experiment! What is a bioinformatics experiment? You need to know your data/input sources You need to understand your methods and their assumptions You need a plan to get from point
Proxer 7 Manager szoftver felhasználói leírás
 Proxer 7 Manager szoftver felhasználói leírás A program az induláskor elkezdi keresni az eszközöket. Ha van olyan eszköz, amely virtuális billentyűzetként van beállítva, akkor azokat is kijelzi. Azokkal
Proxer 7 Manager szoftver felhasználói leírás A program az induláskor elkezdi keresni az eszközöket. Ha van olyan eszköz, amely virtuális billentyűzetként van beállítva, akkor azokat is kijelzi. Azokkal
Miskolci Egyetem Gazdaságtudományi Kar Üzleti Információgazdálkodási és Módszertani Intézet. Correlation & Linear. Petra Petrovics.
 Correlation & Linear Regression in SPSS Petra Petrovics PhD Student Types of dependence association between two nominal data mixed between a nominal and a ratio data correlation among ratio data Exercise
Correlation & Linear Regression in SPSS Petra Petrovics PhD Student Types of dependence association between two nominal data mixed between a nominal and a ratio data correlation among ratio data Exercise
Contact us Toll free (800) fax (800)
 fax (800)") Table of Contents Thank you for purchasing our product, your business is greatly appreciated. If you have any questions, comments, or concerns with the product you received please contact the factory.
Table of Contents Thank you for purchasing our product, your business is greatly appreciated. If you have any questions, comments, or concerns with the product you received please contact the factory.
STUDENT LOGBOOK. 1 week general practice course for the 6 th year medical students SEMMELWEIS EGYETEM. Name of the student:
 STUDENT LOGBOOK 1 week general practice course for the 6 th year medical students Name of the student: Dates of the practice course: Name of the tutor: Address of the family practice: Tel: Please read
STUDENT LOGBOOK 1 week general practice course for the 6 th year medical students Name of the student: Dates of the practice course: Name of the tutor: Address of the family practice: Tel: Please read
A logaritmikus legkisebb négyzetek módszerének karakterizációi
 A logaritmikus legkisebb négyzetek módszerének karakterizációi Csató László laszlo.csato@uni-corvinus.hu MTA Számítástechnikai és Automatizálási Kutatóintézet (MTA SZTAKI) Operációkutatás és Döntési Rendszerek
A logaritmikus legkisebb négyzetek módszerének karakterizációi Csató László laszlo.csato@uni-corvinus.hu MTA Számítástechnikai és Automatizálási Kutatóintézet (MTA SZTAKI) Operációkutatás és Döntési Rendszerek
3. MINTAFELADATSOR KÖZÉPSZINT. Az írásbeli vizsga időtartama: 30 perc. III. Hallott szöveg értése
 Oktatáskutató és Fejlesztő Intézet TÁMOP-3.1.1-11/1-2012-0001 XXI. századi közoktatás (fejlesztés, koordináció) II. szakasz ANGOL NYELV 3. MINTAFELADATSOR KÖZÉPSZINT Az írásbeli vizsga időtartama: 30 perc
Oktatáskutató és Fejlesztő Intézet TÁMOP-3.1.1-11/1-2012-0001 XXI. századi közoktatás (fejlesztés, koordináció) II. szakasz ANGOL NYELV 3. MINTAFELADATSOR KÖZÉPSZINT Az írásbeli vizsga időtartama: 30 perc
SQL/PSM kurzorok rész
 SQL/PSM kurzorok --- 2.rész Tankönyv: Ullman-Widom: Adatbázisrendszerek Alapvetés Második, átdolgozott kiadás, Panem, 2009 9.3. Az SQL és a befogadó nyelv közötti felület (sormutatók) 9.4. SQL/PSM Sémában
SQL/PSM kurzorok --- 2.rész Tankönyv: Ullman-Widom: Adatbázisrendszerek Alapvetés Második, átdolgozott kiadás, Panem, 2009 9.3. Az SQL és a befogadó nyelv közötti felület (sormutatók) 9.4. SQL/PSM Sémában
INDEXSTRUKTÚRÁK III.
 2MU05_Bitmap.pdf camü_ea INDEXSTRUKTÚRÁK III. Molina-Ullman-Widom: Adatbázisrendszerek megvalósítása Panem, 2001könyv 5.4. Bittérkép indexek fejezete alapján Oracle: Indexek a gyakorlatban Oracle Database
2MU05_Bitmap.pdf camü_ea INDEXSTRUKTÚRÁK III. Molina-Ullman-Widom: Adatbázisrendszerek megvalósítása Panem, 2001könyv 5.4. Bittérkép indexek fejezete alapján Oracle: Indexek a gyakorlatban Oracle Database
7 th Iron Smelting Symposium 2010, Holland
 7 th Iron Smelting Symposium 2010, Holland Október 13-17 között került megrendezésre a Hollandiai Alphen aan den Rijn városában található Archeon Skanzenben a 7. Vasolvasztó Szimpózium. Az öt napos rendezvényen
7 th Iron Smelting Symposium 2010, Holland Október 13-17 között került megrendezésre a Hollandiai Alphen aan den Rijn városában található Archeon Skanzenben a 7. Vasolvasztó Szimpózium. Az öt napos rendezvényen
Rezgésdiagnosztika. Diagnosztika 02 --- 1
 Rezgésdiagnosztika Diagnosztika 02 --- 1 Diagnosztika 02 --- 2 A rezgéskép elemzésével kimutatható gépészeti problémák Minden gép, mely tartalmaz forgó részt (pl. motor, generátor, szivattyú, ventilátor,
Rezgésdiagnosztika Diagnosztika 02 --- 1 Diagnosztika 02 --- 2 A rezgéskép elemzésével kimutatható gépészeti problémák Minden gép, mely tartalmaz forgó részt (pl. motor, generátor, szivattyú, ventilátor,
Klaszterezés, 2. rész
 Klaszterezés, 2. rész Csima Judit BME, VIK, Számítástudományi és Információelméleti Tanszék 208. április 6. Csima Judit Klaszterezés, 2. rész / 29 Hierarchikus klaszterezés egymásba ágyazott klasztereket
Klaszterezés, 2. rész Csima Judit BME, VIK, Számítástudományi és Információelméleti Tanszék 208. április 6. Csima Judit Klaszterezés, 2. rész / 29 Hierarchikus klaszterezés egymásba ágyazott klasztereket
Expression analysis of PIN genes in root tips and nodules of Lotus japonicus
 Article Expression analysis of PIN genes in root tips and nodules of Lotus japonicus Izabela Sańko-Sawczenko 1, Dominika Dmitruk 1, Barbara Łotocka 1, Elżbieta Różańska 1 and Weronika Czarnocka 1, * 1
Article Expression analysis of PIN genes in root tips and nodules of Lotus japonicus Izabela Sańko-Sawczenko 1, Dominika Dmitruk 1, Barbara Łotocka 1, Elżbieta Różańska 1 and Weronika Czarnocka 1, * 1
MATEMATIKA ANGOL NYELVEN MATHEMATICS
 ÉRETTSÉGI VIZSGA 2005. május 10. MATEMATIKA ANGOL NYELVEN MATHEMATICS EMELT SZINTŰ ÍRÁSBELI VIZSGA HIGHER LEVEL WRITTEN EXAMINATION Az írásbeli vizsga időtartama: 240 perc Time allowed for the examination:
ÉRETTSÉGI VIZSGA 2005. május 10. MATEMATIKA ANGOL NYELVEN MATHEMATICS EMELT SZINTŰ ÍRÁSBELI VIZSGA HIGHER LEVEL WRITTEN EXAMINATION Az írásbeli vizsga időtartama: 240 perc Time allowed for the examination:
Miskolci Egyetem Gazdaságtudományi Kar Üzleti Információgazdálkodási és Módszertani Intézet Factor Analysis
 Factor Analysis Factor analysis is a multiple statistical method, which analyzes the correlation relation between data, and it is for data reduction, dimension reduction and to explore the structure. Aim
Factor Analysis Factor analysis is a multiple statistical method, which analyzes the correlation relation between data, and it is for data reduction, dimension reduction and to explore the structure. Aim
Create & validate a signature
 IOTA TUTORIAL 7 Create & validate a signature v.0.0 KNBJDBIRYCUGVWMSKPVA9KOOGKKIRCBYHLMUTLGGAV9LIIPZSBGIENVBQ9NBQWXOXQSJRIRBHYJ9LCTJLISGGBRFRTTWD ABBYUVKPYFDJWTFLICYQQWQVDPCAKNVMSQERSYDPSSXPCZLVKWYKYZMREAEYZOSPWEJLHHFPYGSNSUYRZXANDNQTTLLZA
IOTA TUTORIAL 7 Create & validate a signature v.0.0 KNBJDBIRYCUGVWMSKPVA9KOOGKKIRCBYHLMUTLGGAV9LIIPZSBGIENVBQ9NBQWXOXQSJRIRBHYJ9LCTJLISGGBRFRTTWD ABBYUVKPYFDJWTFLICYQQWQVDPCAKNVMSQERSYDPSSXPCZLVKWYKYZMREAEYZOSPWEJLHHFPYGSNSUYRZXANDNQTTLLZA
Word and Polygon List for Obtuse Triangular Billiards II
 Word and Polygon List for Obtuse Triangular Billiards II Richard Evan Schwartz August 19, 2008 Abstract This is the list of words and polygons we use for our paper. 1 Notation To compress our notation
Word and Polygon List for Obtuse Triangular Billiards II Richard Evan Schwartz August 19, 2008 Abstract This is the list of words and polygons we use for our paper. 1 Notation To compress our notation
Where in the Genome does Replication Begin?
 Where in the Genome does Replication Begin? Chapter 1 Bioinformatics Algorithms Phillip Compaeu and Pavel Pevzner ebook link: https://stepic.org/course/bioinformatics-algorithms-2/ DNA Replication Replication
Where in the Genome does Replication Begin? Chapter 1 Bioinformatics Algorithms Phillip Compaeu and Pavel Pevzner ebook link: https://stepic.org/course/bioinformatics-algorithms-2/ DNA Replication Replication
Where in the Genome does Replication Begin?
 Where in the Genome does Replication Begin? Chapter 1 Bioinformatics Algorithms Phillip Compaeu and Pavel Pevzner ebook link: http://beta.stepic.org/2 It has not escaped our notice that the specific pairing
Where in the Genome does Replication Begin? Chapter 1 Bioinformatics Algorithms Phillip Compaeu and Pavel Pevzner ebook link: http://beta.stepic.org/2 It has not escaped our notice that the specific pairing
CLUSTALW Multiple Sequence Alignment
 Version 3.2 CLUSTALW Multiple Sequence Alignment Selected Sequences) FETA_GORGO FETA_HORSE FETA_HUMAN FETA_MOUSE FETA_PANTR FETA_RAT Import Alignments) Return Help Report Bugs Fasta label *) Workbench
Version 3.2 CLUSTALW Multiple Sequence Alignment Selected Sequences) FETA_GORGO FETA_HORSE FETA_HUMAN FETA_MOUSE FETA_PANTR FETA_RAT Import Alignments) Return Help Report Bugs Fasta label *) Workbench
Unit 10: In Context 55. In Context. What's the Exam Task? Mediation Task B 2: Translation of an informal letter from Hungarian to English.
 Unit 10: In Context 55 UNIT 10 Mediation Task B 2 Hungarian into English In Context OVERVIEW 1. Hungarian and English in Context 2. Step By Step Exam Techniques Real World Link Students who have studied
Unit 10: In Context 55 UNIT 10 Mediation Task B 2 Hungarian into English In Context OVERVIEW 1. Hungarian and English in Context 2. Step By Step Exam Techniques Real World Link Students who have studied
EN United in diversity EN A8-0206/419. Amendment
 22.3.2019 A8-0206/419 419 Article 2 paragraph 4 point a point i (i) the identity of the road transport operator; (i) the identity of the road transport operator by means of its intra-community tax identification
22.3.2019 A8-0206/419 419 Article 2 paragraph 4 point a point i (i) the identity of the road transport operator; (i) the identity of the road transport operator by means of its intra-community tax identification
A modern e-learning lehetőségei a tűzoltók oktatásának fejlesztésében. Dicse Jenő üzletfejlesztési igazgató
 A modern e-learning lehetőségei a tűzoltók oktatásának fejlesztésében Dicse Jenő üzletfejlesztési igazgató How to apply modern e-learning to improve the training of firefighters Jenő Dicse Director of
A modern e-learning lehetőségei a tűzoltók oktatásának fejlesztésében Dicse Jenő üzletfejlesztési igazgató How to apply modern e-learning to improve the training of firefighters Jenő Dicse Director of
Utolsó frissítés / Last update: február Szerkesztő / Editor: Csatlós Árpádné
 Utolsó frissítés / Last update: 2016. február Szerkesztő / Editor: Csatlós Árpádné TARTALOM / Contents BEVEZETŐ / Introduction... 2 FELNŐTT TAGBÉLYEGEK / Adult membership stamps... 3 IFJÚSÁGI TAGBÉLYEGEK
Utolsó frissítés / Last update: 2016. február Szerkesztő / Editor: Csatlós Árpádné TARTALOM / Contents BEVEZETŐ / Introduction... 2 FELNŐTT TAGBÉLYEGEK / Adult membership stamps... 3 IFJÚSÁGI TAGBÉLYEGEK
USER MANUAL Guest user
 USER MANUAL Guest user 1 Welcome in Kutatótér (Researchroom) Top menu 1. Click on it and the left side menu will pop up 2. With the slider you can make left side menu visible 3. Font side: enlarging font
USER MANUAL Guest user 1 Welcome in Kutatótér (Researchroom) Top menu 1. Click on it and the left side menu will pop up 2. With the slider you can make left side menu visible 3. Font side: enlarging font
(c) 2004 F. Estrada & A. Jepson & D. Fleet Canny Edges Tutorial: Oct. 4, '03 Canny Edges Tutorial References: ffl imagetutorial.m ffl cannytutorial.m
 2004 F. Estrada & A. Jepson & D. Fleet Canny Edges Tutorial: Oct. 4, '03 Canny Edges Tutorial References: ffl imagetutorial.m ffl cannytutorial.m") Canny Edges Tutorial: Oct. 4, '03 Canny Edges Tutorial References: ffl imagetutorial.m ffl cannytutorial.m ffl ~jepson/pub/matlab/isetoolbox/tutorials ffl ~jepson/pub/matlab/utvistoolbox/tutorials ffl
Canny Edges Tutorial: Oct. 4, '03 Canny Edges Tutorial References: ffl imagetutorial.m ffl cannytutorial.m ffl ~jepson/pub/matlab/isetoolbox/tutorials ffl ~jepson/pub/matlab/utvistoolbox/tutorials ffl
ELEKTRONIKAI ALAPISMERETEK ANGOL NYELVEN
 ÉRETTSÉGI VIZSGA 2013. május 23. ELEKTRONIKAI ALAPISMERETEK ANGOL NYELVEN KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA 2013. május 23. 8:00 Az írásbeli vizsga időtartama: 180 perc Pótlapok száma Tisztázati Piszkozati EMBERI
ÉRETTSÉGI VIZSGA 2013. május 23. ELEKTRONIKAI ALAPISMERETEK ANGOL NYELVEN KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA 2013. május 23. 8:00 Az írásbeli vizsga időtartama: 180 perc Pótlapok száma Tisztázati Piszkozati EMBERI
MATEMATIKA ANGOL NYELVEN
 ÉRETTSÉGI VIZSGA 2016. október 18. MATEMATIKA ANGOL NYELVEN KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA 2016. október 18. 8:00 I. Időtartam: 45 perc Pótlapok száma Tisztázati Piszkozati EMBERI ERŐFORRÁSOK MINISZTÉRIUMA
ÉRETTSÉGI VIZSGA 2016. október 18. MATEMATIKA ANGOL NYELVEN KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA 2016. október 18. 8:00 I. Időtartam: 45 perc Pótlapok száma Tisztázati Piszkozati EMBERI ERŐFORRÁSOK MINISZTÉRIUMA
Statistical Dependence
 Statistical Dependence Petra Petrovics Statistical Dependence Deinition: Statistical dependence exists when the value o some variable is dependent upon or aected by the value o some other variable. Independent
Statistical Dependence Petra Petrovics Statistical Dependence Deinition: Statistical dependence exists when the value o some variable is dependent upon or aected by the value o some other variable. Independent
16F628A megszakítás kezelése
 16F628A megszakítás kezelése A 'megszakítás' azt jelenti, hogy a program normális, szekvenciális futása valamilyen külső hatás miatt átmenetileg felfüggesztődik, és a vezérlést egy külön rutin, a megszakításkezelő
16F628A megszakítás kezelése A 'megszakítás' azt jelenti, hogy a program normális, szekvenciális futása valamilyen külső hatás miatt átmenetileg felfüggesztődik, és a vezérlést egy külön rutin, a megszakításkezelő
Bioinformatics: Blending. Biology and Computer Science
 Bioinformatics: Blending Biology and Computer Science MDNMSITNTPTSNDACLSIVHSLMCHRQ GGESETFAKRAIESLVKKLKEKKDELDSL ITAITTNGAHPSKCVTIQRTLDGRLQVAG RKGFPHVIYARLWRWPDLHKNELKHVK YCQYAFDLKCDSVCVNPYHYERVVSPGI DLSGLTLQSNAPSSMMVKDEYVHDFEG
Bioinformatics: Blending Biology and Computer Science MDNMSITNTPTSNDACLSIVHSLMCHRQ GGESETFAKRAIESLVKKLKEKKDELDSL ITAITTNGAHPSKCVTIQRTLDGRLQVAG RKGFPHVIYARLWRWPDLHKNELKHVK YCQYAFDLKCDSVCVNPYHYERVVSPGI DLSGLTLQSNAPSSMMVKDEYVHDFEG
ANGOL NYELV KÖZÉPSZINT SZÓBELI VIZSGA I. VIZSGÁZTATÓI PÉLDÁNY
 ANGOL NYELV KÖZÉPSZINT SZÓBELI VIZSGA I. VIZSGÁZTATÓI PÉLDÁNY A feladatsor három részből áll 1. A vizsgáztató társalgást kezdeményez a vizsgázóval. 2. A vizsgázó egy szituációs feladatban vesz részt a
ANGOL NYELV KÖZÉPSZINT SZÓBELI VIZSGA I. VIZSGÁZTATÓI PÉLDÁNY A feladatsor három részből áll 1. A vizsgáztató társalgást kezdeményez a vizsgázóval. 2. A vizsgázó egy szituációs feladatban vesz részt a
2. Local communities involved in landscape architecture in Óbuda
 Év Tájépítésze pályázat - Wallner Krisztina 2. Közösségi tervezés Óbudán Óbuda jelmondata: Közösséget építünk, ennek megfelelően a formálódó helyi közösségeket bevonva fejlesztik a közterületeket. Békásmegyer-Ófaluban
Év Tájépítésze pályázat - Wallner Krisztina 2. Közösségi tervezés Óbudán Óbuda jelmondata: Közösséget építünk, ennek megfelelően a formálódó helyi közösségeket bevonva fejlesztik a közterületeket. Békásmegyer-Ófaluban
MATEMATIKA ANGOL NYELVEN
 ÉRETTSÉGI VIZSGA 2014. május 6. MATEMATIKA ANGOL NYELVEN EMELT SZINTŰ ÍRÁSBELI VIZSGA 2014. május 6. 8:00 Az írásbeli vizsga időtartama: 240 perc Pótlapok száma Tisztázati Piszkozati EMBERI ERŐFORRÁSOK
ÉRETTSÉGI VIZSGA 2014. május 6. MATEMATIKA ANGOL NYELVEN EMELT SZINTŰ ÍRÁSBELI VIZSGA 2014. május 6. 8:00 Az írásbeli vizsga időtartama: 240 perc Pótlapok száma Tisztázati Piszkozati EMBERI ERŐFORRÁSOK
Cashback 2015 Deposit Promotion teljes szabályzat
 Cashback 2015 Deposit Promotion teljes szabályzat 1. Definitions 1. Definíciók: a) Account Client s trading account or any other accounts and/or registers maintained for Számla Az ügyfél kereskedési számlája
Cashback 2015 Deposit Promotion teljes szabályzat 1. Definitions 1. Definíciók: a) Account Client s trading account or any other accounts and/or registers maintained for Számla Az ügyfél kereskedési számlája
Trinucleotide Repeat Diseases: CRISPR Cas9 PacBio no PCR Sequencing MFMER slide-1
 Trinucleotide Repeat Diseases: CRISPR Cas9 PacBio no PCR Sequencing 2015 MFMER slide-1 Fuch s Eye Disease TCF 4 gene Fuchs occurs in about 4% of the US population. Leads to deteriorating vision without
Trinucleotide Repeat Diseases: CRISPR Cas9 PacBio no PCR Sequencing 2015 MFMER slide-1 Fuch s Eye Disease TCF 4 gene Fuchs occurs in about 4% of the US population. Leads to deteriorating vision without
Bevezetés a kvantum-informatikába és kommunikációba 2015/2016 tavasz
 Bevezetés a kvantum-informatikába és kommunikációba 2015/2016 tavasz Kvantumkapuk, áramkörök 2016. március 3. A kvantummechanika posztulátumai (1-2) 1. Állapotleírás Zárt fizikai rendszer aktuális állapota
Bevezetés a kvantum-informatikába és kommunikációba 2015/2016 tavasz Kvantumkapuk, áramkörök 2016. március 3. A kvantummechanika posztulátumai (1-2) 1. Állapotleírás Zárt fizikai rendszer aktuális állapota
KIEGÉSZÍTŽ FELADATOK. Készlet Bud. Kap. Pápa Sopr. Veszp. Kecsk. 310 4 6 8 10 5 Pécs 260 6 4 5 6 3 Szomb. 280 9 5 4 3 5 Igény 220 200 80 180 160
 KIEGÉSZÍTŽ FELADATOK (Szállítási probléma) Árut kell elszállítani három telephelyr l (Kecskemét, Pécs, Szombathely) öt területi raktárba, melyek Budapesten, Kaposváron, Pápán, Sopronban és Veszprémben
KIEGÉSZÍTŽ FELADATOK (Szállítási probléma) Árut kell elszállítani három telephelyr l (Kecskemét, Pécs, Szombathely) öt területi raktárba, melyek Budapesten, Kaposváron, Pápán, Sopronban és Veszprémben
Csima Judit április 9.
 Osztályozókról még pár dolog Csima Judit BME, VIK, Számítástudományi és Információelméleti Tanszék 2018. április 9. Csima Judit Osztályozókról még pár dolog 1 / 19 SVM (support vector machine) ez is egy
Osztályozókról még pár dolog Csima Judit BME, VIK, Számítástudományi és Információelméleti Tanszék 2018. április 9. Csima Judit Osztályozókról még pár dolog 1 / 19 SVM (support vector machine) ez is egy
Efficient symmetric key private authentication
 Efficient symmetric key private authentication Cryptographic Protocols (EIT ICT MSc) Dr. Levente Buttyán Associate Professor BME Hálózati Rendszerek és Szolgáltatások Tanszék Lab of Cryptography and System
Efficient symmetric key private authentication Cryptographic Protocols (EIT ICT MSc) Dr. Levente Buttyán Associate Professor BME Hálózati Rendszerek és Szolgáltatások Tanszék Lab of Cryptography and System
Tutorial 1 The Central Dogma of molecular biology
 oday DN RN rotein utorial 1 he entral Dogma of molecular biology Information flow in genetics:» ranscription» ranslation» Making sense of genomic information Information content in DN - Information content
oday DN RN rotein utorial 1 he entral Dogma of molecular biology Information flow in genetics:» ranscription» ranslation» Making sense of genomic information Information content in DN - Information content
István Micsinai Csaba Molnár: Analysing Parliamentary Data in Hungarian
 István Micsinai Csaba Molnár: Analysing Parliamentary Data in Hungarian The Hungarian Comparative Agendas Project Participant of international Comparative Agendas Project Datasets on: Laws (1949-2014)
István Micsinai Csaba Molnár: Analysing Parliamentary Data in Hungarian The Hungarian Comparative Agendas Project Participant of international Comparative Agendas Project Datasets on: Laws (1949-2014)
Utolsó frissítés / Last update: Szeptember / September Szerkesztő / Editor: Csatlós Árpádné
 Utolsó frissítés / Last update: 2018. Szeptember / September Szerkesztő / Editor: Csatlós Árpádné TARTALOM / Contents BEVEZETŐ / Introduction... 2 FELNŐTT TAGBÉLYEGEK / Adult membership stamps... 3 IFJÚSÁGI
Utolsó frissítés / Last update: 2018. Szeptember / September Szerkesztő / Editor: Csatlós Árpádné TARTALOM / Contents BEVEZETŐ / Introduction... 2 FELNŐTT TAGBÉLYEGEK / Adult membership stamps... 3 IFJÚSÁGI
Lopocsi Istvánné MINTA DOLGOZATOK FELTÉTELES MONDATOK. (1 st, 2 nd, 3 rd CONDITIONAL) + ANSWER KEY PRESENT PERFECT + ANSWER KEY
 + ANSWER KEY PRESENT PERFECT + ANSWER KEY") Lopocsi Istvánné MINTA DOLGOZATOK FELTÉTELES MONDATOK (1 st, 2 nd, 3 rd CONDITIONAL) + ANSWER KEY PRESENT PERFECT + ANSWER KEY FELTÉTELES MONDATOK 1 st, 2 nd, 3 rd CONDITIONAL I. A) Egészítsd ki a mondatokat!
Lopocsi Istvánné MINTA DOLGOZATOK FELTÉTELES MONDATOK (1 st, 2 nd, 3 rd CONDITIONAL) + ANSWER KEY PRESENT PERFECT + ANSWER KEY FELTÉTELES MONDATOK 1 st, 2 nd, 3 rd CONDITIONAL I. A) Egészítsd ki a mondatokat!
Basic Arrays. Contents. Chris Wild & Steven Zeil. May 28, Description 3
 Chris Wild & Steven Zeil May 28, 2013 Contents 1 Description 3 1 2 Example 4 3 Tips 6 4 String Literals 7 4.1 Description...................................... 7 4.2 Example........................................
Chris Wild & Steven Zeil May 28, 2013 Contents 1 Description 3 1 2 Example 4 3 Tips 6 4 String Literals 7 4.1 Description...................................... 7 4.2 Example........................................
ELEKTRONIKAI ALAPISMERETEK ANGOL NYELVEN
 ÉRETTSÉGI VIZSGA 2008. május 26. ELEKTRONIKAI ALAPISMERETEK ANGOL NYELVEN KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA 2008. május 26. 8:00 Az írásbeli vizsga időtartama: 180 perc Pótlapok száma Tisztázati Piszkozati OKTATÁSI
ÉRETTSÉGI VIZSGA 2008. május 26. ELEKTRONIKAI ALAPISMERETEK ANGOL NYELVEN KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA 2008. május 26. 8:00 Az írásbeli vizsga időtartama: 180 perc Pótlapok száma Tisztázati Piszkozati OKTATÁSI
Dense Matrix Algorithms (Chapter 8) Alexandre David B2-206
 Alexandre David B2-206") Dense Matrix Algorithms (Chapter 8) Alexandre David B2-206 Dense Matrix Algorithm Dense or full matrices: few known zeros. Other algorithms for sparse matrix. Square matrices for pedagogical purposes only
Dense Matrix Algorithms (Chapter 8) Alexandre David B2-206 Dense Matrix Algorithm Dense or full matrices: few known zeros. Other algorithms for sparse matrix. Square matrices for pedagogical purposes only
MATEMATIKA ANGOL NYELVEN
 ÉRETTSÉGI VIZSGA MATEMATIKA ANGOL NYELVEN EMELT SZINTŰ ÍRÁSBELI VIZSGA 8:00 Az írásbeli vizsga időtartama: 240 perc Pótlapok száma Tisztázati Piszkozati OKTATÁSI ÉS KULTURÁLIS MINISZTÉRIUM Matematika angol
ÉRETTSÉGI VIZSGA MATEMATIKA ANGOL NYELVEN EMELT SZINTŰ ÍRÁSBELI VIZSGA 8:00 Az írásbeli vizsga időtartama: 240 perc Pótlapok száma Tisztázati Piszkozati OKTATÁSI ÉS KULTURÁLIS MINISZTÉRIUM Matematika angol
KN-CP50. MANUAL (p. 2) Digital compass. ANLEITUNG (s. 4) Digitaler Kompass. GEBRUIKSAANWIJZING (p. 10) Digitaal kompas
 Digital compass. ANLEITUNG (s. 4) Digitaler Kompass. GEBRUIKSAANWIJZING (p. 10) Digitaal kompas") KN-CP50 MANUAL (p. ) Digital compass ANLEITUNG (s. 4) Digitaler Kompass MODE D EMPLOI (p. 7) Boussole numérique GEBRUIKSAANWIJZING (p. 0) Digitaal kompas MANUALE (p. ) Bussola digitale MANUAL DE USO (p.
KN-CP50 MANUAL (p. ) Digital compass ANLEITUNG (s. 4) Digitaler Kompass MODE D EMPLOI (p. 7) Boussole numérique GEBRUIKSAANWIJZING (p. 0) Digitaal kompas MANUALE (p. ) Bussola digitale MANUAL DE USO (p.
Probabilistic Analysis and Randomized Algorithms. Alexandre David B2-206
 Probabilistic Analysis and Randomized Algorithms Alexandre David B2-206 Today Counting. Basic probability. Appendix C Introduction to randomized algorithms. Chapter 5 27-10-2006 AA1 2 Counting Rule of
Probabilistic Analysis and Randomized Algorithms Alexandre David B2-206 Today Counting. Basic probability. Appendix C Introduction to randomized algorithms. Chapter 5 27-10-2006 AA1 2 Counting Rule of
Directors and Officers Liability Insurance Questionnaire Adatlap vezetõ tisztségviselõk és felügyelõbizottsági tagok felelõsségbiztosításához
 Directors and Officers Liability Insurance Questionnaire Adatlap vezetõ tisztségviselõk és felügyelõbizottsági tagok felelõsségbiztosításához 1. Name, legal form and address of company Társaság neve, címe,
Directors and Officers Liability Insurance Questionnaire Adatlap vezetõ tisztségviselõk és felügyelõbizottsági tagok felelõsségbiztosításához 1. Name, legal form and address of company Társaság neve, címe,
ENROLLMENT FORM / BEIRATKOZÁSI ADATLAP
 ENROLLMENT FORM / BEIRATKOZÁSI ADATLAP CHILD S DATA / GYERMEK ADATAI PLEASE FILL IN THIS INFORMATION WITH DATA BASED ON OFFICIAL DOCUMENTS / KÉRJÜK, TÖLTSE KI A HIVATALOS DOKUMENTUMOKBAN SZEREPLŐ ADATOK
ENROLLMENT FORM / BEIRATKOZÁSI ADATLAP CHILD S DATA / GYERMEK ADATAI PLEASE FILL IN THIS INFORMATION WITH DATA BASED ON OFFICIAL DOCUMENTS / KÉRJÜK, TÖLTSE KI A HIVATALOS DOKUMENTUMOKBAN SZEREPLŐ ADATOK
First experiences with Gd fuel assemblies in. Tamás Parkó, Botond Beliczai AER Symposium 2009.09.21 25.
 First experiences with Gd fuel assemblies in the Paks NPP Tams Parkó, Botond Beliczai AER Symposium 2009.09.21 25. Introduction From 2006 we increased the heat power of our units by 8% For reaching this
First experiences with Gd fuel assemblies in the Paks NPP Tams Parkó, Botond Beliczai AER Symposium 2009.09.21 25. Introduction From 2006 we increased the heat power of our units by 8% For reaching this
Local fluctuations of critical Mandelbrot cascades. Konrad Kolesko
 Local fluctuations of critical Mandelbrot cascades Konrad Kolesko joint with D. Buraczewski and P. Dyszewski Warwick, 18-22 May, 2015 Random measures µ µ 1 µ 2 For given random variables X 1, X 2 s.t.
Local fluctuations of critical Mandelbrot cascades Konrad Kolesko joint with D. Buraczewski and P. Dyszewski Warwick, 18-22 May, 2015 Random measures µ µ 1 µ 2 For given random variables X 1, X 2 s.t.
(NGB_TA024_1) MÉRÉSI JEGYZŐKÖNYV
 MÉRÉSI JEGYZŐKÖNYV") Kommunikációs rendszerek programozása (NGB_TA024_1) MÉRÉSI JEGYZŐKÖNYV (5. mérés) SIP telefonközpont készítése Trixbox-szal 1 Mérés helye: Széchenyi István Egyetem, L-1/7 laboratórium, 9026 Győr, Egyetem
Kommunikációs rendszerek programozása (NGB_TA024_1) MÉRÉSI JEGYZŐKÖNYV (5. mérés) SIP telefonközpont készítése Trixbox-szal 1 Mérés helye: Széchenyi István Egyetem, L-1/7 laboratórium, 9026 Győr, Egyetem
Tudományos Ismeretterjesztő Társulat
 Sample letter number 3. Russell Ltd. 57b Great Hawthorne Industrial Estate Hull East Yorkshire HU 19 5BV 14 Bebek u. Budapest H-1105 10 December, 2009 Ref.: complaint Dear Sir/Madam, After seeing your
Sample letter number 3. Russell Ltd. 57b Great Hawthorne Industrial Estate Hull East Yorkshire HU 19 5BV 14 Bebek u. Budapest H-1105 10 December, 2009 Ref.: complaint Dear Sir/Madam, After seeing your
Descriptive Statistics
 Descriptive Statistics Petra Petrovics DESCRIPTIVE STATISTICS Definition: Descriptive statistics is concerned only with collecting and describing data Methods: - statistical tables and graphs - descriptive
Descriptive Statistics Petra Petrovics DESCRIPTIVE STATISTICS Definition: Descriptive statistics is concerned only with collecting and describing data Methods: - statistical tables and graphs - descriptive
Tavaszi Sporttábor / Spring Sports Camp. 2016. május 27 29. (péntek vasárnap) 27 29 May 2016 (Friday Sunday)
 27 29 May 2016 (Friday Sunday)") Tavaszi Sporttábor / Spring Sports Camp 2016. május 27 29. (péntek vasárnap) 27 29 May 2016 (Friday Sunday) SZÁLLÁS / ACCOMODDATION on a Hotel Gellért*** szálloda 2 ágyas szobáiban, vagy 2x2 ágyas hostel
Tavaszi Sporttábor / Spring Sports Camp 2016. május 27 29. (péntek vasárnap) 27 29 May 2016 (Friday Sunday) SZÁLLÁS / ACCOMODDATION on a Hotel Gellért*** szálloda 2 ágyas szobáiban, vagy 2x2 ágyas hostel
Választási modellek 3
 Választási modellek 3 Prileszky István Doktori Iskola 2018 http://www.sze.hu/~prile Forrás: A Self Instructing Course in Mode Choice Modeling: Multinomial and Nested Logit Models Prepared For U.S. Department
Választási modellek 3 Prileszky István Doktori Iskola 2018 http://www.sze.hu/~prile Forrás: A Self Instructing Course in Mode Choice Modeling: Multinomial and Nested Logit Models Prepared For U.S. Department
Intézményi IKI Gazdasági Nyelvi Vizsga
 Intézményi IKI Gazdasági Nyelvi Vizsga Név:... Születési hely:... Születési dátum (év/hó/nap):... Nyelv: Angol Fok: Alapfok 1. Feladat: Olvasáskészséget mérő feladat 20 pont Olvassa el a szöveget és válaszoljon
Intézményi IKI Gazdasági Nyelvi Vizsga Név:... Születési hely:... Születési dátum (év/hó/nap):... Nyelv: Angol Fok: Alapfok 1. Feladat: Olvasáskészséget mérő feladat 20 pont Olvassa el a szöveget és válaszoljon
Alternating Permutations
 Alternating Permutations Richard P. Stanley M.I.T. Definitions A sequence a 1, a 2,..., a k of distinct integers is alternating if a 1 > a 2 < a 3 > a 4 a 3
Alternating Permutations Richard P. Stanley M.I.T. Definitions A sequence a 1, a 2,..., a k of distinct integers is alternating if a 1 > a 2 < a 3 > a 4 a 3
University of Bristol - Explore Bristol Research
 Al-Salihi, S. A. A., Scott, T. A., Bailey, A. M., & Foster, G. D. (2017). Improved vectors for Agrobacterium mediated genetic manipulation of Hypholoma spp. and other homobasidiomycetes. Journal of Microbiological
Al-Salihi, S. A. A., Scott, T. A., Bailey, A. M., & Foster, G. D. (2017). Improved vectors for Agrobacterium mediated genetic manipulation of Hypholoma spp. and other homobasidiomycetes. Journal of Microbiological
ELEKTRONIKAI ALAPISMERETEK ANGOL NYELVEN
 ÉRETTSÉGI VIZSGA 2012. május 25. ELEKTRONIKAI ALAPISMERETEK ANGOL NYELVEN KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA 2012. május 25. 8:00 Az írásbeli vizsga időtartama: 180 perc Pótlapok száma Tisztázati Piszkozati NEMZETI
ÉRETTSÉGI VIZSGA 2012. május 25. ELEKTRONIKAI ALAPISMERETEK ANGOL NYELVEN KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA 2012. május 25. 8:00 Az írásbeli vizsga időtartama: 180 perc Pótlapok száma Tisztázati Piszkozati NEMZETI
MATEMATIKA ANGOL NYELVEN
 ÉRETTSÉGI VIZSGA 2014. október 14. MATEMATIKA ANGOL NYELVEN KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA 2014. október 14. 8:00 I. Időtartam: 45 perc Pótlapok száma Tisztázati Piszkozati EMBERI ERŐFORRÁSOK MINISZTÉRIUMA
ÉRETTSÉGI VIZSGA 2014. október 14. MATEMATIKA ANGOL NYELVEN KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA 2014. október 14. 8:00 I. Időtartam: 45 perc Pótlapok száma Tisztázati Piszkozati EMBERI ERŐFORRÁSOK MINISZTÉRIUMA
Kvantum-informatika és kommunikáció 2015/2016 ősz. A kvantuminformatika jelölésrendszere szeptember 11.
 Kvantum-informatika és kommunikáció 2015/2016 ősz A kvantuminformatika jelölésrendszere 2015. szeptember 11. Mi lehet kvantumbit? Kvantum eszközök (1) 15=5 3 Bacsárdi Képek forrása: IBM's László, Almaden
Kvantum-informatika és kommunikáció 2015/2016 ősz A kvantuminformatika jelölésrendszere 2015. szeptember 11. Mi lehet kvantumbit? Kvantum eszközök (1) 15=5 3 Bacsárdi Képek forrása: IBM's László, Almaden
SAJTÓKÖZLEMÉNY Budapest 2011. július 13.
 SAJTÓKÖZLEMÉNY Budapest 2011. július 13. A MinDig TV a legdinamikusabban bıvülı televíziós szolgáltatás Magyarországon 2011 elsı öt hónapjában - A MinDig TV Extra a vezeték nélküli digitális televíziós
SAJTÓKÖZLEMÉNY Budapest 2011. július 13. A MinDig TV a legdinamikusabban bıvülı televíziós szolgáltatás Magyarországon 2011 elsı öt hónapjában - A MinDig TV Extra a vezeték nélküli digitális televíziós
Lexington Public Schools 146 Maple Street Lexington, Massachusetts 02420
 146 Maple Street Lexington, Massachusetts 02420 Surplus Printing Equipment For Sale Key Dates/Times: Item Date Time Location Release of Bid 10/23/2014 11:00 a.m. http://lps.lexingtonma.org (under Quick
146 Maple Street Lexington, Massachusetts 02420 Surplus Printing Equipment For Sale Key Dates/Times: Item Date Time Location Release of Bid 10/23/2014 11:00 a.m. http://lps.lexingtonma.org (under Quick
MATEMATIKA ANGOL NYELVEN MATHEMATICS
 ÉRETTSÉGI VIZSGA 2005. május 10. MATEMATIKA ANGOL NYELVEN MATHEMATICS KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA STANDARD LEVEL WRITTEN EXAMINATION I. Időtartam: 45 perc Time allowed: 45 minutes Number of extra sheets
ÉRETTSÉGI VIZSGA 2005. május 10. MATEMATIKA ANGOL NYELVEN MATHEMATICS KÖZÉPSZINTŰ ÍRÁSBELI VIZSGA STANDARD LEVEL WRITTEN EXAMINATION I. Időtartam: 45 perc Time allowed: 45 minutes Number of extra sheets
MATEMATIKA ANGOL NYELVEN
 ÉRETTSÉGI VIZSGA 2012. május 8. MATEMATIKA ANGOL NYELVEN EMELT SZINTŰ ÍRÁSBELI VIZSGA 2012. május 8. 8:00 Az írásbeli vizsga időtartama: 240 perc Pótlapok száma Tisztázati Piszkozati NEMZETI ERŐFORRÁS
ÉRETTSÉGI VIZSGA 2012. május 8. MATEMATIKA ANGOL NYELVEN EMELT SZINTŰ ÍRÁSBELI VIZSGA 2012. május 8. 8:00 Az írásbeli vizsga időtartama: 240 perc Pótlapok száma Tisztázati Piszkozati NEMZETI ERŐFORRÁS
Rotary District 1911 DISTRICT TÁMOGATÁS IGÉNYLŐ LAP District Grants Application Form
 1 A Future Vision pilot célja a Future Vision Plan (Jövőkép terv) egyszerűsített támogatási modelljének tesztelése, és a Rotaristák részvételének növelése a segélyezési folyamatokban. A teszt során a districteknek
1 A Future Vision pilot célja a Future Vision Plan (Jövőkép terv) egyszerűsített támogatási modelljének tesztelése, és a Rotaristák részvételének növelése a segélyezési folyamatokban. A teszt során a districteknek
Hogyan használja az OROS online pótalkatrész jegyzéket?
 Hogyan használja az OROS online pótalkatrész jegyzéket? Program indítása/program starts up Válassza ki a weblap nyelvét/choose the language of the webpage Látogasson el az oros.hu weboldalra, majd klikkeljen
Hogyan használja az OROS online pótalkatrész jegyzéket? Program indítása/program starts up Válassza ki a weblap nyelvét/choose the language of the webpage Látogasson el az oros.hu weboldalra, majd klikkeljen
Cloud computing. Cloud computing. Dr. Bakonyi Péter.
 Cloud computing Cloud computing Dr. Bakonyi Péter. 1/24/2011 1/24/2011 Cloud computing 2 Cloud definició A cloud vagy felhő egy platform vagy infrastruktúra Az alkalmazások és szolgáltatások végrehajtására
Cloud computing Cloud computing Dr. Bakonyi Péter. 1/24/2011 1/24/2011 Cloud computing 2 Cloud definició A cloud vagy felhő egy platform vagy infrastruktúra Az alkalmazások és szolgáltatások végrehajtására
Adattípusok. Max. 2GByte
 Adattípusok Típus Méret Megjegyzés Konstans BIT 1 bit TRUE/FALSE SMALLINT 2 byte -123 INTEGER 4 byte -123 COUNTER 4 byte Automatikus 123 REAL 4 byte -12.34E-2 FLOAT 8 byte -12.34E-2 CURRENCY / MONEY 8
Adattípusok Típus Méret Megjegyzés Konstans BIT 1 bit TRUE/FALSE SMALLINT 2 byte -123 INTEGER 4 byte -123 COUNTER 4 byte Automatikus 123 REAL 4 byte -12.34E-2 FLOAT 8 byte -12.34E-2 CURRENCY / MONEY 8
Dynamic freefly DIVE POOL LINES
 Dynamic freefly 3.rész Part 3 DIVE POOL LINES A dynamic repülés három fajta mozgásból áll: Dynamic freeflying is composed of three types of movements: -Lines (vízszintes "szlalom") (horizontal "slalom")
Dynamic freefly 3.rész Part 3 DIVE POOL LINES A dynamic repülés három fajta mozgásból áll: Dynamic freeflying is composed of three types of movements: -Lines (vízszintes "szlalom") (horizontal "slalom")