Mellékhatások és minőségi hibák kombinációs előfordulása
|
|
|
- Júlia Faragó
- 9 évvel ezelőtt
- Látták:
Átírás
1 Mellékhatások és minőségi hibák kombinációs előfordulása Dr. Oláh Attila, Dr. Mészáros Márta DEBRECENI EGÉSZSÉGÜGYI MINŐSÉGÜGYI NAPOK (DEMIN XIV.) Debrecen május

2 Miről is lesz szó Jogszabályi háttér Általános elvek Minőségi reklamációk és mellékhatások MedDRA EudraVigilance adatbázis 2

3 Európai jogszabályok: Jogszabályi háttér Vonatkozó EU-s és magyar jogszabályok Az Európai Parlament és a Tanács 726/2004/EK rendelete (2004. március 31.) az emberi, illetve állatgyógyászati felhasználásra szánt gyógyszerek engedélyezésére és felügyeletére vonatkozó közösségi eljárások meghatározásáról és az Európai Gyógyszerügynökség létrehozásáról Az Európai Parlament és a Tanács 1235/2010/EU rendelete (2010. december 15.) az emberi, illetve állatgyógyászati felhasználásra szánt gyógyszerek engedélyezésére és felügyeletére vonatkozó közösségi eljárások meghatározásáról és az Európai Gyógyszerügynökség létrehozásáról szóló 726/2004/EK rendeletnek és a fejlett terápiás gyógyszerkészítményekről szóló 1394/2007/EK rendeletnek az emberi felhasználásra szánt gyógyszerekkel összefüggésben követendő farmakovigilancia tekintetében történő módosításáról Az Európai Parlament és a Tanács 2001/83/EK irányelve (2001. november 6.) az emberi felhasználásra szánt gyógyszerek közösségi kódexéről Az Európai Parlament és a Tanács 2010/84/EU irányelve (2010. december 15.) az emberi felhasználásra szánt gyógyszerek közösségi kódexéről szóló 2001/83/EK európai parlamenti és tanácsi irányelvnek a farmakovigilancia tekintetében történő módosításáról Az Európai Parlament és a Tanács 2001/20/EK irányelve (2001. április 4.) az emberi felhasználásra szánt gyógyszerkészítményekkel végzett klinikai vizsgálatok során alkalmazandó helyes klinikai gyakorlat bevezetésére vonatkozó tagállami törvényi, rendeleti és közigazgatási rendelkezések közelítéséről 3 Magyar jogszabályok: évi XCV. Törvény az emberi alkalmazásra kerülő gyógyszerekről és egyéb, a gyógyszerpiacot szabályozó törvények módosításáról 15/2012. (VIII.22.) EMMI rendelet az emberi alkalmazásra kerülő gyógyszerek farmakovigilanciájáról 35/2005. (VIII. 26.) EüM rendelet az emberi felhasználásra kerülő vizsgálati készítmények klinikai vizsgálatáról és a helyes klinikai gyakorlat alkalmazásáról

4 Mi is pontosan a minőségi hiba vagy minőségi kifogás? Röviden: a felhasználó által tapasztalt minőségbeli eltérés, valamint a gyártással, csomagolással és használattal összefüggésbe hozható, észlelt potenciális hiba. Példák minőségi kifogásokra Megváltozott a termék megjelenése Nem kielégítő adagolás Hiányzó alkotórészek Szennyezett termék Sérült termék Azonosítási hiba Termék hiányosságok Csomagolási hiba pl.: krémes állagú termék vízszerűvé vált pl.: orrspray, ami nem kellően porlaszt pl.: aerosol inhaláló szájadéka hiányzik pl.: idegen anyag a termékben vagy terméken (pl. penész) pl.: törött applikátor pl.: krémek dobozán kenőcs felirat pl.: hiányoznak tabletták az üvegcséből pl.: olvashatatlan lejárati idő a fiolán 4

5 Minimálisan szükséges információk a jelentéshez Minőségi kifogás Nemkívánatos esemény /mellékhatás Termék pontos neve (hatáserősség, kiszerelés is) Gyártási szám és lejárati idő Minőségi kifogás leírása Magát a kifogásolt termék ne dobjuk el! Termék pontos neve (hatáserősség, kiszerelés, gyártási szám és lejárati idő is) Beteg adatai (monogram, nem, kor) Nemkívánatos esemény leírása: Maga a gyógyszeralkalmazás ténye is nemkívánatos esemény, tehát önmagában, egyéb tünetek híján is jelentendő Bármi, a gyógyszerhasználattal együtt járó tünet, lelet, esemény Bejelentő adatai (név, foglalkozás, elérhetőség) 5

6 Mi történhet a kifogásolt termékek használatakor? Számos esetben a minőségi probléma nem akadályozza meg automatikusan a termék alkalmazását. A felhasználó figyelmét elkerülhetik bizonyos problémák (pl. nem biztos, hogy a használó érzékeli a nem kellő mértékű porlasztást, ha nincs vonatkozási alapja, nem mindig látványos, ha esetleg idegen anyag van a termékben, vagy a rosszul olvasható lejárati idő egy már lejárt termék alkalmazásához vezethet). Ezekben az esetekben a gyógyszer alkalmazásra kerülhet, ami egy minőségileg nem megfelelő termék bejutását jelentheti a páciens szervezetébe. Egy nem megfelelően porlasztó orrspray esetében ezt túlvagy aluldozírozáshoz vezethet. Idegen anyag pl. egy infúziós palackban fertőzést okozhat. Egy, a lejárati idő után alkalmazott termék esetében pedig a gyártó már nem szavatolhatja, hogy a hatóanyag kifejti a kellő hatást. A lényeg: onnantól kezdve, hogy a beteg kapcsolatba kerül egy nem megfelelő minőségű termékkel, már nem csak a termék hibáját szükséges felismerni, és megfelelő módon kivizsgálni, hanem a gyógyszer hatásának kitett pácienst is vizsgálni kell abból a szempontból, hogy kifejtett-e, és ha igen, akkor milyen nemkívánatos hatást fejtett ki a betegnél a kifogásolt gyógyszer vagy termék alkalmazása. 6
. Ezekben az esetekben a gyógyszer alkalmazásra kerülhet, ami egy minőségileg nem megfelelő termék bejutását jelentheti a páciens szervezetébe.")
7 Mit kell tenni akkor, ha a kifogásolt gyógyszer alkalmazásra került? Abban az esetben, ha minőségileg nem megfelelő termék került felhasználásra, vagy a készítmény esetleg nem megfelelő módon került alkalmazásra, és ebből adódtak problémák, mind a termék, mind pedig a beteg vagy páciens szemszögéből meg kell vizsgálni a problémát. 2 irányban kell folytatni a vizsgálódást, egyrészt maga a minőségi probléma, vagyis a termék, illetve emellett a beteg is kivizsgálásra kell, hogy kerüljön. Mindez természetesen nem egymástól elkülönülve történik. Közvetve vagy közvetlenül a termék forgalmazóját mindenképpen be kell vonni a folyamatba, aki a hatósággal karöltve az egymással összefüggő panaszt és eseményt alaposan kivizsgálja és értékeli. 7

8 8
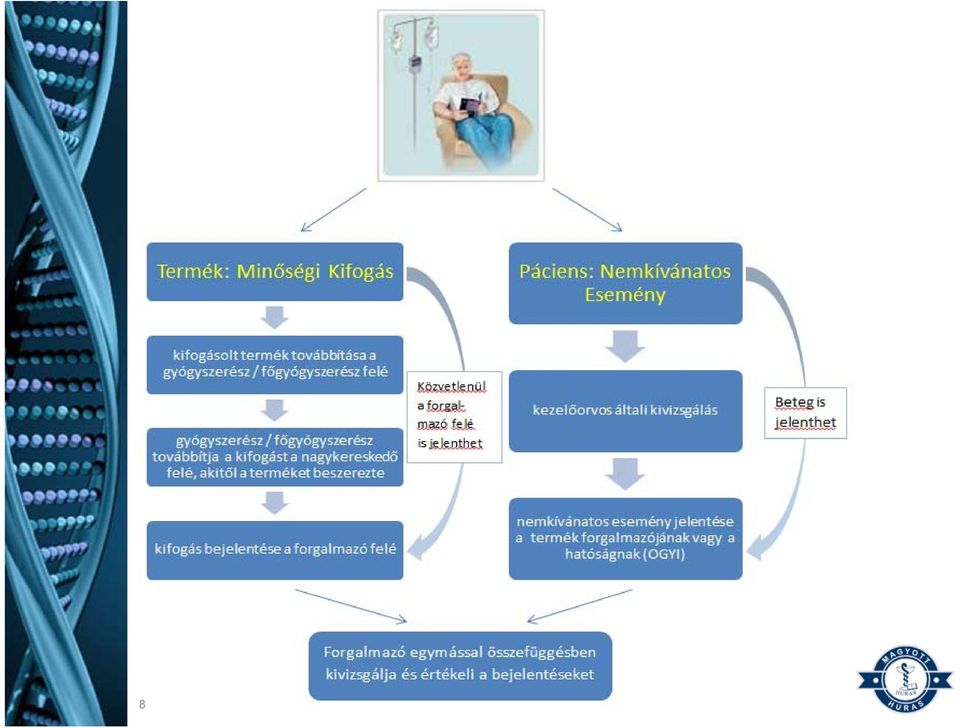
9 Mi az, ami nem tekinthető minőségi kifogásnak? Vannak olyan termékkel kapcsolatos felhasználói visszajelzések is, amelyek nem tekinthetőek minőségi kifogásnak, ilyenek például a következők: Fogyasztói visszajelzések, amelyek nem minősülnek minőségi kifogásnak: A felhasználó elégedetlen a termék megjelenésével, konstrukciójával, árával, stb. A termék használatát MEGELŐZŐEN, annak használatával kapcsolatban felmerülő kérdések A terméke elérhetőségével kapcsolatos kérdések, panaszok 9

10 Definíciók Mellékhatás: a gyógyszerek által kiváltott káros és nem kívánt hatás. Mellékhatásnak minősülnek a gyógyszerek szokásos adagolása során a forgalomba hozatali engedély szerinti alkalmazásból eredő káros és nem kívánt hatásokon kívül a gyógyszerelési hibából, valamint a forgalomba hozatali engedélyben nem szereplő felhasználásból eredő káros, nem kívánt hatások is, beleértve a gyógyszer helytelen használatát és az azzal való visszaélést; Nem várt mellékhatás: olyan mellékhatás, amelynek jellege, súlyossága vagy kimenetele nem egyezik meg az alkalmazási előírásban felsorolt mellékhatásokkal; Súlyos mellékhatás: olyan mellékhatás, amely az életet veszélyezteti, kórházi kezelést tesz szükségessé, illetve azt meghosszabbítja, maradandó vagy jelentős egészségkárosodást, rokkantságot, veleszületett rendellenességet, születési hibát vagy halált okoz; 10

11 Forgalombahozatali ingedély jogosult kötelezettsége Fenntartani egy rendszert, amely biztosítja a piacon elhelyezett termékért vállalt felelősségét és kötelezettségeit Súlyos mellékhatások gyorsított jelentése (7/15 nap) Nem súlyos nemkívánatos reakciók (mellékhatások) jelentése (90 nap) 11
 Nem súlyos nemkívánatos reakciók (mellékhatások)")
12 4 minimum információ, spontán/klinika, súlyosság Bejövő jelentés TRIAGE Egyedi gyógyszebiztonsági jelentések általános workflow Regisztráció Teljes adatbevitel (beleértve narratív írás), MedDRA kódolás Adatbevitel + ellenőrzés ( QC lépés) Ok-okozati összefüggés értékelés, vártság/listázottság eldöntése Orvosi értékelés és végső jóváhagyás ICH E2B XML (EV-WEB / Gateway) v. CIOMS ( , fax, posta) 12 Jelentés
 v.")
13 Medical Dictionary for Regulatory Activities MedDRA

14 MedDRA (Medical Dictionary for Regulatory Activities) Klinikailag validált nemzetközi orvosi terminológia, mely a gyógyszerfejlesztés összes fázisára vonatkozik, kivéve az állati toxikológiát ICH (International Conference on Harmonisation) dolgozta ki Karbantartását, disztribúcióját az MSSO (Maintenance and Support Services Organization) végzi jan. óta kötelező EU-ban a nemkívánatos reakciók jelentésénél ( gyógyszerbiztonsági információk könnyebb megosztása és elemzése) Évente kétszer (márc./szept.) kerül frissítésre új MedDRA verzió/alverzió kiadásával 14
 Évente kétszer (márc.")
15 A terminológia hatásköre MedDRA Terminológiában foglaltak: jelek tünetek betegségek diagnózisok terápiás indikációk ideértve a jeleket, tüneteket, betegségeket, diagnózisokat, a betegség diagnózisát vagy profilaxisát és a fiziológiás funkció módosítását vizsgálatok nevei és minőségi eredményei pl. növekedett, csökkent, normális, abnormális, kimutatható, nem kimutatható, pozitív és negatív gyógyszerelési hibákra és termékminőségre vonatkozó kifejezések műtéti és orvosi eljárások orvosi/szociális/családi anamnézis Hatáskörön kívül esnek: gyógyszer/termék terminológia berendezés/eszköz/diagnosztikai termék terminológia vizsgálat felépítése demográfia minősítők, melyek a populációkra vonatkoznak és nem az egyes betegekre (pl. ritka, gyakori) laboratóriumi paraméterekhez társuló numerikus értékek (pl. szérum nátrium 141 meq/l). súlyosságot leíró kifejezések. Az olyan leírók mint súlyos és enyhe csak akkor használatosak, ha a kifejezés specifikusságára vonatkoznak (pl. súlyos kontra enyhe mentalis retardatio). 15

16 Szerkezet, hierarchia MedDRA Rendszer szerv osztály (SOC) [26] Magas-szintű csoport kifejezés (HLGT) Magas-szintű kifejezés (HLT) Preferált kifejezés (PT) [> 20 ezer] 16 Legalacsonyabb szintű kifejezés (LLT) [> 71 ezer] kódolás Jelentett kifejezés (RT)
![(HLT) Preferált kifejezés (PT) [> 20 ezer] 16 Legalacsonyabb](/docs-images/47/16609332/images/page_16.jpg "szintű kifejezés (LLT) [> 71 ezer] kódolás Jelentett kifejezés")
17 MedDRA Rendszer szerv osztályok (System Organ Classes) Általános betegségek és beadás helyi feltételek Bőr és subcutan szövet betegségek Cardialis betegségek Endokrin betegségek Fertőzések és infestatiók Fül és és labhyrintus betegségek Gastrointestinalis betegségek Hepatobiliáris betegségek Idegrendszeri betegségek Immunrendszer betegségek Légzési, mellkasi és mediastinal betegségek Metabolizmus és táplálkozási betegségek Musculoskeletalis és kötőszövet betegségek Neoplazmák jóindulatú, rosszindulatú ésmeghatározhatatlan (beleértve a cisztákat és a polypokat) Pszichiátriai betegségek Reproduktív rendszer és emlő betegségek Sebészeti és orvosi beavatkozások Sérülés, mérgezés és beavatkozási komplikációk Szem betegségek Társadalmi körülmények Terhesség, gyermekágy és perinatalis állapotok Vaszkularis betegségek Vele született, családi és genetikai betegségek Vér- és lymphatikus rendszer betegségek Vese és húgyúti betegségek Vizsgálatok 17

18

19 Kódolás MedDRA Kódolási irányelvek Points to Consider dokumentum Minden bejelentett kifejezéshez a bejelentő szavait legpontosabban tükröző érvényes MedDRA LLT-t kell kiválasztani A kódolás helyességének és következetességnek az ellenőrzése Tilos új orvosi fogalmat vagy információt hozzáadni a kódolandó kifejezéshez Nem szabad diagnózist felállítani a bejelentő által megnevezett tünetekből (ha a diagnózist nem jelentette)! 19

20 Points to Consider (PTC) Két PTC dokumentum: Term Selection Data Retrieval and Presentation MedDRA A MedDRA egy nagyon fontos eszköz a közös nyelv és a harmonizáció szempontjából 20

21 MedDRA Term Selection: Points to Consider ICH Irányító Testületének egyik munkacsoportja fejlesztette ki Hatóságok és ipar képviselőivel EU, Japán, USA Kanadai megfigyelő, MSSO, JMO ICH-endorsed guide MedDRA felhasználók részére Megfelelő (orvosi) kifejezés kiválasztáshoz nyújt segítséget az ipar és a hatóság számára Cél: előmozdítani a megfelelő és következetes kódolást, illetve a megosztott adatok egységes értelmezését Évente kétszer kerül frissítésre minden egyes új MedDRA verzió/alverzió publikálásával 21
22 EUDRAVIGILANCE
23 EudraVigilance adatbázis 2001-ben lépett először működésbe lényege, hogy az Európai Unión belül a gyógyszer mellékhatás bejelentések áramlása a hatóságok és a gyógyszeripar között elektronikus úton történik az ICH E2B standardok alkalmazásával. Moduljai: EudraVigilance Clinical Trial Module (EVCTM) Suspected Unexpected Serious Adverse Reaction (SUSAR) riportok elektronikus fogadására Directive 2001/20/EC rendelkezéseinek megfelelően EudraVigilance Post-Authorisation Module (EVPM) spontán egyedi mellékhatás jelentések ( post-authorisation ICSR ) elektronikus fogadására Regulation (EC) No 726/2004, Directive 2001/83/EC rendelkezéseinek megfelelően a klinikai vizsgálatokból származó SUSAR jelentéseket» május 1. óta (EVCTM), a spontán mellékhatás bejelentéseket» november 20. óta (EVPM) kell elektronikus úton jelenteni az EMA rendelkezései értelmében 23
24
25 EudraVigilance Data Analysis System (EVDAS) Miből is épül fel? Source systems 25
26 Source systems - Forrás rendszerek Ezek a rendszerek fogadják és tárolják azon adatokat, melyeket EudraVigilance (EV)-be jelentettek, ill. EV használ. A következőket foglalja magába: EudraVigilance Database Management system (EVDBMS), EudraVigilance Medicinal Product Dictionary (EVMPD) EudraVigilance registration database (MAH, EudraCT), Medical Dictionary for Regulatory Affairs (MedDRA) és más nemzetközileg elfogadott és elismert standard terminológiák melyet a EudraVigilance használ (pl. European Pharmacopoeia Dosage Forms). Az EDVBMS az alábbi adatokat tartalmazza: PM-ICSRs: Individual Case Safety Reports EudraVigilance Post-Authorisation Module (EVPM) CT-ICSRs (SUSARs): Individual Case Safety Reports EudraVigilance Clinical Trial module (EVCTM) PSUR-ICSRs: Individual Case Safety Reports from Periodic Safety Update Reports EVPM ASR-ICSRs: Individual Case Safety Reports from Annual Safety Reports EVCTM BACKLOG-ICSRs: Individual Case Safety Reports (retrospective!) EVPM or EVCTM 26
27 ETL & EV Data Warehouse & Data Analysis toolkit EV Data Warehouse Adattárház, mely különféle szűrések/lekérdezések, táblázatok elkészítéshez van optimalizálva. Az itt lévő adatok az ETL révén esténként (naponta!) frissülnek az adott napi új információkkal EV Data Analysis toolkit Előre definiált táblák, grafikonok a lekérdezett adatok minél jobb prezentálása végett. Testre szabható lekérdezések. Extraction, Transfer and Loading Azon folyamatok, szabályok összessége, melynek során az adatok átkerülnek a forrás adatbázisokból az adattárházba
28 KÖSZÖNÖM SZÉPEN A FIGYELMET!